The Longevity FAQ
Table of Contents
NOTE: This was written in 2020, and if I were to write it again I would probably do differently, add more structure, add some more sections. I also don't have that much time to update this FAQ, so there are other papers worth checking out written post-2020 that will not be here.
Inasmuch as one enjoys being alive, waiting longer until the signs of frailty and old age occur seems an appealing proposition, and so there is an entire field of research dedicated to understand the aging process. A recent summary for a popular audience is in David Sinclair's recent book Lifespan. But I wanted to provide a deeper and more concise explanation, plus communicating not only the results but also their robustness. There is also a previous Longevity FAQ from Laura Deming, but I thought something a bit longer that explains the field from the ground up should exist.
At first, reading about research regarding longevity can seem like magic: "We knocked out Sirt1 in mice, leading to reduced lifespan". That sentence is not only compressing a lot of information (What does it mean to knock out? What's Sirt1?) but also once we know that knocking out Sirt1 means to stop a gene from being expressed (i.e. stopping the cell from manufacturing the protein associated with that gene), we may want to know things like "Are there different ways of knocking out genes? How do different genes related in the genetics of aging relate to each other? If we do the same things in dogs, does it work?"
My goal here is to demystify what seems initially obscure, and to make available a summary of the current state of the art, the quality of the evidence available so far, and what promising avenues of research are being pursued at the moment.
If you disagree with anything said here, you can get in touch with me if you want to correct this FAQ (You may get paid if you do so!) or to suggest additions to it. To write this FAQ, I've reviewed over 100 papers, most of these literature reviews themselves. Everything I say is referenced, but you can find the sources, tagged by area here. As people comment and review the FAQ, I'll be making changes to make sure it stays up to date.
If you want a more conceptual introduction to aging, read this first.
Molecular Biology 101 and assorted concepts
I will explain first some key concepts as this will make it easier to understand what is going on
Genes
From a structural perspective, DNA is a double-helix of two chains of nucleotides (adenine, thymine, guanine, cytosine, ATGC). If we take one of the strands of this double-helix, one can identify different sections that do different things, these are the genes. In turn, a given gene can be further broken down into codons, or triplets of nucleotides, e.g. the sequence A-T-G.
Transcription
To go from the sequence of codons to something more interesting, the DNA is copied by the enzyme RNA polymerase into a strand of RNA (pre-mRNA) which is then modified to eventually become messenger RNA (mRNA). A single strand of pre-mRNA can yield many strands of mRNA. A strand of pre-mRNA can be thought of as a chain of "useful" sections (exons) and "separator" sections (introns). pre-mRNA is cut at the introns and joined back together by a spliceosome, a complex association of 80 proteins conforming a molecular machine. This process is not always the same! Depending on how this cutting and pasting happens, different mRNA sequences will result, in turn leading to different proteins being made out of the same gene.
Translation
mRNA is basically like one of the two strands of DNA but instead of using ATGC, it has uracil instead of thymine, so AUGC. The strand of mRNA is sent into the cytoplasm of the cell where they reach the ribosomes. There, yet another form of RNA, transfer RNA (tRNA) reads the mRNA and assembles a chain of amino acids in the order dictated by the codons. So if the mRNA says AUG-GCU-UAA we get methionine-alanine (The last codon is a stop codon, and stops the transcription right there).
But because there are so many of them, and because they have a few functional groups attached to them, they will interact with each other and end up curling up to conform a protein.
Occasionally more than a single chain will be required for a protein to be assembled. To recap, if we write down the DNA sequence, we can know what amino acids will be produced and how they will be stitched together. But knowing how the protein will assemble itself, and what final shape it will adopt is a very hard problem
The narrative outlined above is what is known as the Central dogma of molecular biology The real picture is somewhat more complicated. For example, once a protein is floating around, this protein can interact with the DNA itself and regulate its activity; this is the basis for epigenetics.
Gene Expression regulation
We still need one more thing: Almost every cell in an organism shares the same DNA (There are exceptions, as in the adaptive immune system), yet a neuron is markedly different from a cell in the liver. This is because not every gene is expressed in every cell: they can be switched on and off (gene regulation), by means of either internal (maybe the concentration of some protein, what proteins are present inside) or external (exposition to certain molecules outside of the cell) signals.
This regulation can happen at various levels in the gene -> protein route explained above.
- Before transcription: This is covered under epigenetics, in the next section.
- During transcription: Certain proteins, called transcription factors, can bind to specific genes and promote or repress their transcription into mRNA.
- During translation: One can also interfere with the process that takes mRNA and assembles the amino acids together. For example, one can reduce the lifespan of mRNA in the cell, if there's less of it, less proteins will be made. This is the basis of RNA interference (RNAi), used to knock down or reduce the activity of a gene.
- Post-translation: Even when the route has come to an end and a protein has been assembled, one can still act on these newly assembled proteins, be it by disassembling them, or by tagging them by attaching certain chemical groups to them. An example of this is phosphorylation, where a phosphoryl group is attached to a protein. Phosphorylation - or any other modification to a protein - doesn't always have the same effect, it may increase the activity of a protein, or reduce it, depending on what the protein is. Another ubiquitous mechanism is ubiquitination, sticking small ubiquitin proteins to other proteins; proteins so tagged are flagged for the cell to recycle them, reducing the concentration of the protein in the cytoplasm, leading to reduced activity.
Epigenetics
So far we have considered DNA as something that's just sitting there, like a file in a hard drive from which we only read. But it's possible to interpret this genetic code in different ways, and this is the main mechanism by which cells end up being so different.
The DNA double helix is spun around histones, another kind of protein (To make things more convoluted, there are different kinds of histones). This wrapped DNA plus some other ancillary proteins form together the chromatin molecule. Both the nucleotides itself and the histones can have their chemical structure altered without having the sequence of nucleotides changed, and these changes affect what genes get expressed -this is like writing meta-data to our source file. In the case of DNA, one way is sticking methyl groups to the DNA nucleotides, which typically will stop the gene from even being transcribed. The histones that are part of chromatin can also be methylated, though here this mark can act both as a promoter or repressor of gene expression. Other forms of epigenetic modifications are possible, like (de)acetylation, similar to methylation but involving an acetyl group, which typically opens up the chromatin facilitating gene expression; we'll see in a moment a class of proteins, the sirtuin, that are directly involved in this process and are relevant to the aging process.
Another mode of epigenetic regulation of gene expression is the formation of heterochromatin: DNA is not floating around with all of its nucleotides accessible to the transcription machinery; rather it sometimes curls in a tightly packed form -heterochromatin- that inhibits transcription.
It seems like the relevance of epigenetics to aging is higher than it was thought years ago. (Pal and Tyler (2016), Kane and Sinclair, (2019)), and David Sinclair in particular defends a theory of aging that puts it at the core of the issue.
Aging has an associated change in the methylation patterns of our DNA, and this seems similar across individuals, enabling the construction of methylation clocks (Or epigenetic clocks), pioneered by Steve Horvath, which can infer biological age from a readout of the epigenome (Wagner, 2019). The more advanced of these clocks like GrimAge (Lu et al. 2019) are able to predict remaining years of lifespan, or time to the onset of cardiovascular disease.
You can read more about epigenetic clocks in this longer explainer.
Chromosomes and telomeres
One might think that inside the cellular nucleus there are the very visible and nicely shaped chromosomes that we see in textbooks. But this is not true: They are generally very hard to see as they are in an uncoiled state, facilitating their interaction with other proteins. It is only when the cell is about to divide that they become tightly packed.
At the end of the DNA molecule there are telomeres, sections of repeated nucleotides (In humans, TTAGGG). In turn at the end of the telomere there is a 'knot' or loop around the shelterin protein complex. What shelterin (Palm & de Lange, 2008) does is to hide (Or shelter hah) the end of the telomere from the DNA repair machinery: DNA looks like a continuous strand for most of its length except at the ends, so it is efficient for the cell to recognise 'ends' of this strand as breaks, and try to repair them. But of course you wouldn't want that in the end of the chromosome, because it's a legitimate feature. However, if true damage happens to the telomeric section, it's not going to be repaired, so telomeres steadily accumulate damage. If shelterin is removed, the cell recognises this as a large DNA damage point and commits cellular suicide (apoptosis) or enters replicative arrest (senescence). We'll get to senescent cells later.
When the cell divides and chromosomes are assembled, telomeres end up as 'caps' in the arms of the chromosomes, but when the DNA is being replicated, because of the way the enzyme DNA polymerase (Which does the replication job) works, it fails to copy a few nucleotides at the end, so with each cell replication, the telomeres shorten by about 50 base pairs (bp).
Once the telomere is short enough, it won't be able to maintain the 'loop' structure at its end, thus it will be recognised as a DNA break, triggering cell death. Thus given an initial telomeric length and a rate of attrition per cell division, one could calculate how many times a cell can replicate. This is known as the Hayflick limit.
It's important to remark that rarely the cell will actually reach a point where there is no telomeres left at all: at birth, telomeres are around 10 kilobases long and every year we lose around 25 base pairs (Shammas, 2012) so in theory a cell has enough telomeres to go for 400 years, assuming that there is no other damage to telomeres! (Actually less than that, because there is a critically low telomere length, ~ 2 kb for humans at which the cell triggers cell death)
Cells however induce apoptosis or senescence way before it gets to this point, so while running out of telomere is the ultimate failsafe, it's more common for the telomere to have accumulated so much damage that this suffices to trigger cell death. Thus telomeres act as instruments to measure damage in the cell in general (Victorelli & Passos, 2017).
It's important to note that cell death or senescence will happen when the shorter telomere is shorter than some threshold. For some time much attention was paid to average telomere length, but better measurement techniques made it clearer that it wasn't that way.
However, we do have some cells that replicate a lot (Like the stem cells that produce red blood cells) and their telomeres are not long enough to live long enough through our lives. So for those cells that do need to replicate there is an reverse transcriptase (It makes DNA from RNA, while regular transcriptase does the opposite) enzyme, telomerase, that can lengthen the telomeres. Telomerase is made up of two core proteins, those encoded by the TERT and TERC (An RNA template) genes. The fact that telomerase exists is the reason why sometimes one may read that 'X healthy habit lengthen telomeres', technically it is possible for telomerase to lead to longer telomeres in the population of some cells like red blood cells or leukocytes (The ones typically used to measure telomere length). However, for the rest of the cells, telomere length can only ever shorten. (With the rare exception of the Alternative Lengthening of Telomeres (ALT) mechanism).
Telomeres are useful to suppress cancer: If a cell replicates too fast, it will exhaust their telomeres and die. So typically when cancer does happen it is because telomerase somehow has become active, or more rarely (10% of cases) due to ALT.
Some mutations can cause deficiencies in telomere length and accurate assays can help diagnose this to guide treatment, both in animal models and in humans, mutations in telomerase or abnormalities in the telomeres is linked to disease like pulmonary fibrosis or dyskeratosis congenita (Alder et al. 2018, Lopez-Otin et al., 2013) however for the general population telomere length does not seem very predictive of illness.
To make things more confusing, telomerase seems to help cells continue replicate above and beyond its effect on telomeres (Zhu et al., 1999, Bayat et al., 2020), seemingly having a protective effect of its own.
While the length of telomeres does not predict lifespan across species (e.g. wild-type mice have telomeres that are like ours, not much shorter, while, possible for selection reasons, lab mice have longer telomeres than we do, see Hemann & Greider, 2000), the rate of attrition of telomeres is surprisingly predictive of how long an average individual of a species will live (Whittemore et al., 2019).
Telomere research has shown in the past some problems originated by inaccurate measurement methods (Martin-Ruiz et al. 2014) and low sample size studies (Pepper et al. 2018), which explains this infamous paper from Nobel prize winner Elizabeth Blackburn's lab where chanting “Saa, Taa, Naa, Maa,” elongates your telomeres. Predictably, the largest, properly randomised studies, show no such effect, even when other studies continue to do (Schutte et al., 2020).
All things considered, whether drugs to elongate telomeres will lead to more lifespan or healthspan in the general population remains debated.
The Blasco lab has shown that, without genetic modifications, mice with hyper-long telomeres are more healthy, live longer, and have less incidence of cancer(Munoz-Lorente et al., 2019) . They achieved this, I repeat, without any genetic modification, so telomerase upregulation (Which they also measured) was not the cause here; rather they found a 6x decrease in senescent cells, the effects of which will be discussed in a few sections. This may seem surprising given that we said that telomeres are protection against cancer; but perhaps in this particular case, the cancer effects from longer telomeres are counteracted from the cancer-reducing effects from less inflammation due to reduced cellular senescence (Bonafe et al., 2020).
The opposite also seems true: In zebrafish with very short telomeres, senescence-driven inflammation leads to more cancer (Lex et al., 2020), so while telomere shortening end eventual senescence is a mechanism to control cancer, excessive senescence driven by very short telomeres can induce cancer too!
There is some evidence from Mendelian randomisation studies points to telomere length not being relevant in humans (Kuo et al., 2019.), perhaps because longer telomeres increase the odds of cancer while decreasing those of some other illnesses, for a net effect of zero (Melzer et al. 2019). This is why, while shorter telomeres are sometimes considered a hallmark of aging, not everyone thinks this way (Mather et al. 2010).
DNA replication can occur without the need for telomeres or shortening if the DNA forms a closed loop, as is the case in mitochondrial DNA.
UPDATE (2023-10-17): Check out this new post I wrote on telomeres
Pathways
So we have a bunch of proteins in the cells moving around and bumping into each other. Their shape and molecular composition will influence if they will do anything interesting. For example if you put in a vat some hTERT (The protein made by TERT) and hTR (The RNA made by TERC), they will spontaneously combine to form telomerase. In some cases this reactions require organic catalysts - enzymes - and the enzymes in turn sometimes need other molecules (co-factors) to actually be active.
In cells, there exist defined ways in which proteins react with each other in many ways: Proteins can combine to form larger structures, they can change conformation (shape), react to split into smaller proteins, etc. These usually happen in defined networks, or pathways, which can also feature loops, causing negative or positive feedback, enabling various forms of homeostasis. Here's an example (Pan & Finkel, 2017) of the representation of some of these pathways that also shows their interconectedness. (The -| at the end of an arrow means inhibit, while the -> means promotes)
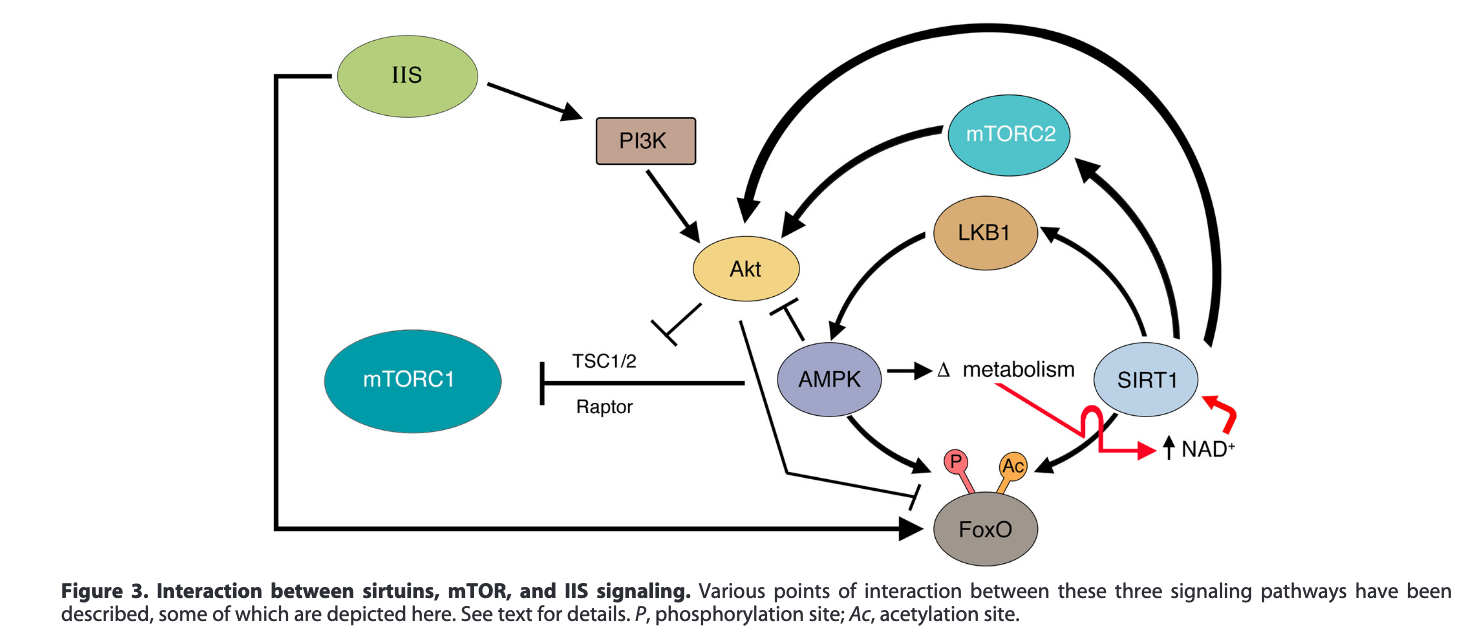
Here's another example (Pal & Tyler, 2016) that also shows the interactions between various drugs or treatments and proteins. The major pathways involved in aging are the insulin / insulin growth factor pathway (Targetable through calorie restriction), mTOR (inhibited by rapamycin), and AMPK (activated by metformin). Note that the diagrams are not exactly the same (Where is LKB1 in the picture below?) This is done to highlight some or other proteins if the author thinks in that particular case one of the intermediate ones does not matter.
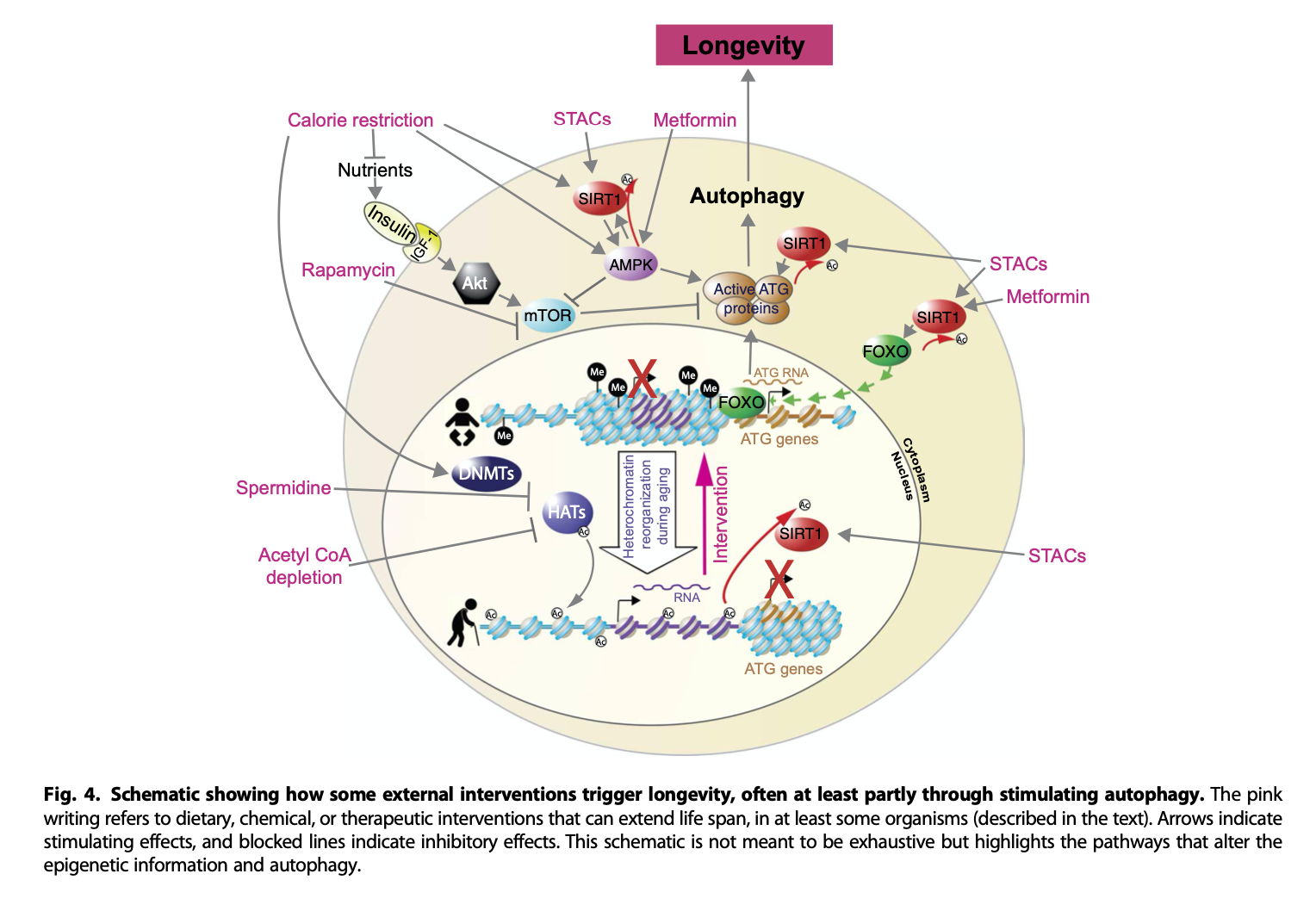
Here's yet another one, from del Cabo et al. (2014)
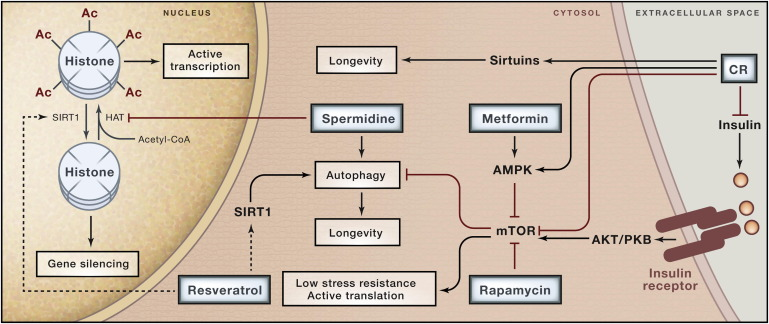
The whole set of pathways is very complex, here you can see a map of the proteins that are involved in regulating longevity, and their interactions.
Dietary restriction is upstream of many of these for good reason: What ties together many of these pathways, which are more or less preserved across species including yeast, is the trade-off between growth and repair. When there is abundance of nutrients, the signal is to focus on reproduction, while when they are scarce, the cell focuses on reducing the production of, and promoting the repair of, damage.
Genome instability
DNA gets damaged over time, but this is surprisingly not as bad as it seems (Vijg & Montagna, 2017).
Damage properly understood includes things like breaks in the double helix, crosslinks or other alterations to the structure of the DNA molecule itself as a carrier of information. Around 100,000 lesions of this kind occur daily on somatic cells, but most of this is usually repaired.
Genome instability refers to an increase in the amount of de novo mutations a given genome experiences. Here, the DNA is not damaged, it's just that the sequence of molecules is different, so there is nothing for the cell to repair; the cell has no way to know (Unless the cell could somehow compare itself to its neighbours) that a given nucleotide is not what it used to. This sounds kind of bad, but most mutations are harmless, as most of the genome is not crucial to the functioning of an organism. The only mutations that are truly related to a decrease in lifespan are those in oncogenes, which lead to cancer (Though note that it has been argued that a weakened immune system might be a more relevant cause (Finn, 2018); the immune system is able to keep cancer cells in check after all. The relevance of this continues to be debated, see Jimenez-Alonso et al. 2018 and Palmer et al., 2018)
You can read a longer review on immunosenescence I wrote here.
Another cause of genome instability is an increase in activity from transposable elements (TEs) like retrotransposons. These guys are sequences that exist in the DNA whose only point in life is to copy themselves and insert themselves back again into the genome. As a result we have >500k copies (17% of our genome!) of a sequences called L1, but 99,9% of them are inactive due to mutations. In maize, 70% of their genome is made of these. However, as aging progresses the repression of the expression of TEs weakens, generating an excess of TE RNA and DNA in the cell, which can trigger an autoimmune response (Bourque et al., 2018, Kane & Sinclair, 2018).
David Sinclair in Lifespan says that DNA damage cannot be that much of an issue in aging because if it were so, cloned animals (From cells of old animals) would be born prematurely aged or otherwise with some defect like shortened lifespan. Furthermore, it has been possible to clone mice in succession over and over to see if mutations accumulate causing premature aging, but this hasn't happened. Elsewhere in the book he says that mice born to older parents tend to present shorter lifespans or are otherwise impaired in some way. The same is true for humans, but Sinclair points to an insufficient genetic reprogramming in the zygote to erase the parental epigenome (which is damaged) rather than DNA mutations, as the cause for these anomalies in the offspring. What's the evidence for this?
In cloned animals (Burgstaller & Brem, 2017) it is true that the story is as Sinclair says, with some caveats: It has not been possible to do successive cloning (cloning an old animal, waiting for that animal to age, then cloning again, and repeat) without limits; no more than 2 in bulls, 5 generations in mice, or 3 generations in pigs, cats, and cattle. It is only by treating zygotes with trichostatin A (Which helps with epigenetic reprogramming) specially designed for this purpose, that 25 successive clonings (At the age of 3 months) have been achieved. Three months however is not that old, lifespan for the strain they used is two years. Given that mice are short-lived anyway (Less time to accumulate mutations), this is not that directly translatable to humans. There is also the very obvious possibility that there are selection effects going on during the cloning process; it's not like they take a single cell and they are able to arbitrarily develop it into a successful clone, the efficiency is very low. Only cells that are relatively damage-free would lead to successful clones. Thus the evidence from the cloning studies is not that strong against a role for DNA damage in aging. To see if they are an issue or not we would need a study where one looks at the correlation between reprogramming efficiency and the levels of DNA damage. As of the writing of this review, that has not been done.
One piece of evidence for DNA damage mattering is that long-lived species have enhanced DNA repair capabilities (Tombline et al., 2019); and it would be unlikely if natural selection has led to this pattern by chance unless all those proteins that are overexpressed in long lived mammals also happen to work through the maintenance of the epigenome with the DNA repair capabilities being merely a happy coincidence. Likewise, the species-specific rate of increase in DNA damage correlates with species lifespan in animals as diverse as flamingos, dolphins, or goats. (Whittemore et al., 2019)
Humans are longer-lived, having more time for mutations to accumulate, and the rate of mutations in humans (germline) are higher than in mice (Lindsay et al., 2019). In turn, the rate of mutations in germline cells is two orders of magnitude less than somatic cells (Milholland et al. 2017), so given enough time, the accumulation of mutations, despite all the redundancies and repair mechanisms, would end up causing problems regardless of the weight one wants to give to DNA damage as a strong causal factor (Niedernhofer et al. 2018). A recent assessment of the evidence (Vijg, 2020) gives more weight to DNA mutations as a main driver of aging, and a more recent study gives evidence against. Short of an in depth review of this particular question, I tentatively side with the "probably is not a major driver of aging" side.
Sirtuins
In 1991, Leonard Guarente and his lab at MIT were doing research on yeast and found a gene that also had an effect on aging (Guarente, 2014): SIR2, which when expressed produces a type of proteins, sirtuins. Upregulating SIR2 leads to longer lifespans up to a point when further upregulation is toxic. Genes analog to SIR2 (orthologs) in species like worms (sir-2.1), flies (dSir2), and mice (SIRT1,SIRT6), have been shown to also increase lifespan (In humans there are seven of these genes, which in turn give rise to seven sirtuin proteins), although some studies dispute some of these results (e.g. for SIRT1, or for SIRT6 in female mice see Sen et al., 2016)
What sirtuins do is to remove acetyl groups from proteins (deacetylation). These acetyl groups or tags in turn moderate the activity of the protein. In particular, sirtuins can deacetylate the histones (in the chromatin), causing the DNA to coil itself back around the histones, in a more stable form than otherwise. (Normally the DNA needs to be unwound to be transcribed, but we don't want it to remain like that forever)
To do their job, sirtuins require the presence of a molecule, coenzyme NAD+, the levels of which decrease as we age. NAD+ plays a role in metabolism as electron carriers during redox reactions. Why does NAD+ decrease? One way is sirtuins consuming it as part of their role in DNA repair; if as we age there are more defects in DNA, then sirtuins is used more often, but in doing so, sirtuins are left without NAD+. In a way, it's shifting from a situation where there is minimal damage and most energy is spent keeping DNA stable to a situation where sirtuins are firefighting all the time, instead of going back to maintain the stability of an already deteriorated DNA. Sinclair, in Lifespan describes their role thus:
When sirtuins shift from their typical priorities to engage in DNA repair, their epigenetic function at home ends for a bit. Then, when the damage is fixed and they head back to home base, they get back to doing what they usually do: controlling genes and making sure the cell retains its identity and optimal function.
But what happens when there’s one emergency after another to tend to? Hurricane after hurricane? Earthquake after earthquake? The repair crews are away from home a lot. The work they normally do piles up. The bills come due, then overdue, and then the folks from collections start calling. The grass grows too long, and soon the president of the neighborhood association is sending nastygrams. The baseball team goes coachless, and the team devolves into the Bad News Bears. And most of all, one of the most important things they do while at home—reproducing—doesn’t get done. This form of hormesis, the original survival circuit, works fine to keep organisms alive in the short term. But unlike longevity molecules that simply mimic hormesis by tweaking sirtuins, mTOR, or AMPK, sending out the troops on fake emergencies, these real emergencies create life-threatening damage.
What could cause so many emergencies? DNA damage. And what causes that? Well, over time, life does. Malign chemicals. Radiation. Even normal DNA copying. These are the things that we’ve come to believe are the causes of aging, but there is a subtle but vital shift we have to make in that manner of thinking. It’s not so much that the sirtuins are overwhelmed, though they probably are when you are sunburned or get an X-ray; what’s happening every day is that the sirtuins and their coworkers that control the epigenome don’t always find their way back to their original gene stations after they are called away. It’s as if a few emergency workers who came to address the damage done in the Gulf Coast by Katrina had lost their home address. Then disaster strikes again and again, and they must redeploy.
Wherever epigenetic factors leave the genome to address damage, genes that should be off, switch on and vice versa. Wherever they stop on the genome, they do the same, altering the epigenome in ways that were never intended when we were born.
Cells lose their identity and malfunction. Chaos ensues. The chaos materializes as aging. This is the epigenetic noise that is at the heart of our unified theory.
Another source of NAD+ repletion is its reaction with the protein CD38, which in old age is thought to become overactive, absorbing increasing amounts of NAD+ (Bonkowski & Sinclair, 2016, Camacho-Pereira, 2016), as well as activation of PARP1, which like sirtuins help with damage repair (Luna et al. 2013). Out of these ones, it is CD38 overactivation, particularly in inflammatory (M1) macrophages that is by far the main culprit of NAD+ depletion (Covarrubias et al., 2019)
There are some other compounds that have been thought to help sirtuins (SIRT-activating compounds, or STACs), one of them being resveratrol (Guarente, 2013); despite the fact that some early studies that found a link between resveratrol and SIRT activity were not done properly, later work does seem to indicate that resveratrol does what it was originally thought to do.
Caloric restriction also seems to have an effect through sirtuins: knocking out SIRT1 in mice that are also caloric restricted does not show any benefits from caloric restriction compared to regular mice regarding lifespan; the same is true for SIRT3 and hearing loss, again in mice.
As one might expect, the results are full of caveats; while overexpressing sirtuins in yeast as we said increases lifespan, with some claiming doesn't happen in flies (Burnett et al., 2011), result that was later challenged by Whitaker et al. (2013) on the grounds that the the gene coding for the sirtuin in flies was excessively overexpressed in the earlier studies, this being toxic. Additionally some have even disputed the yeast results (Lopez-Otin et al., 2013), In mammals,
Regarding mammals, several of the seven mammalian sirtuin paralogs can ameliorate various aspects of aging in mice (Houtkooper et al., 2012; Sebastián et al., 2012). In particular, transgenic overexpression of mammalian SIRT1, which is the closest homolog to invertebrate Sir2, improves aspects of health during aging but does not increase longevity (Herranz et al., 2010). The mechanisms involved in the beneficial effects of SIRT1 are complex and interconnected, including improved genomic stability (Oberdoerffer et al., 2008; Wang et al., 2008) and enhanced metabolic efficiency (Nogueiras et al., 2012) (see “Deregulated Nutrient Sensing”). More compelling evidence for a sirtuin-mediated prolongevity role in mammals has been obtained for SIRT6, which regulates genomic stability, NF-κB signaling, and glucose homeostasis through histone H3K9 deacetylation (Kanfi et al., 2010; Kawahara et al., 2009; Zhong et al., 2010). Mutant mice that are deficient in SIRT6 exhibit accelerated aging (Mostoslavsky et al., 2006), whereas male transgenic mice overexpressing SIRT6 have a longer lifespan than control animals, associated with reduced serum IGF-1 and other indicators of IGF-1 signaling (Kanfi et al., 2012). Interestingly, the mitochondria-located sirtuin SIRT3 has been reported to mediate some of the beneficial effects of dietary restriction (DR) in longevity, though its effects are not due to histone modifications but, rather, due to the deacetylation of mitochondrial proteins (Someya et al., 2010). Very recently, overexpression of SIRT3 has been reported to improve the regenerative capacity of aged hematopoietic stem cells (Brown et al., 2013). Therefore, in mammals, at least three members of the sirtuin family—SIRT1, SIRT3 and SIRT6—contribute to healthy aging.
(Edit 2023-09-04): Here's the latest on sirtuins, from Derek Lowe
But you can still find papers, any number of recently published papers, that assume that the sirtuin longevity story is an accepted biological fact, even when the authors admit that they don’t understand what’s happening and can’t figure out why there hasn’t been more progress. It’s true that “It’s complicated” is a valid response to problems like this in biology. But here’s another valid response that also fits the facts: it’s wrong.
Reactive Oxygen Species (ROS)
We've been talking about damage. One kind of such damage is ROS, the only known molecular species present in the organism capable of cutting off covalent bonds indiscriminately. ROS is produced in substantial amounts at mitochondria as a byproduct of ATP (a molecule used as energy transport) production. ROS can damage among other things telomeres, which leads up to cellular senescence and in turn, aging.
David Sinclair, in Lifespan claims that this is not an issue for aging and that this is the consensus in the field, referring to a theory of aging known as the Oxidative Stress Theory of Aging. Indeed, back in 2009 there was a paper (Perez et al. 2009) proclaiming the theory dead, on the grounds that antioxidant intake does not prolong life, and that deletion of genes that produce antioxidants does not reduce lifespan (Likewise, the addition of extra copies of antioxidant-generating genes did not lengthen lifespan). Likewise the same was said in 2010 of the Mitochondrial Free Radical Theory of Aging or mFRTA (Lapointe, 2010).
Other papers were coming out that were less pessimistic. Salmon et al (2010) admits that ROS may not be involved in lifespan, but that it may be in healthspan, or that the effects may be different (worse) if oxidative stress is chronic rather than acute (As it is in experiments).
In 2013, ROS was shown to be a minor cause of mutations and damage in mtDNA (Kennedy et al. 2013). But that same year, the proponents of the oxidative aging theory replied (Barja, 2013), arguing that the criticisms are unfounded: Antioxidant intake may not lengthen maximum lifespan, but it does increase mean lifespan, longer lived species generate less ROS, additionally the location of ROS generation (In the mitochondria) ensures that extra antioxidants won't get there, so antioxidants may reduce damage outside but not damage to mtDNA. Nor does the theory need the assumption that ROS production increases with age: A constant rate of damage can cause an exponentially increasing mortality rate.
In 2014, an extensive literature review (Stuart et al. 2014) concluded that indeed ROS does not cause aging via indiscriminate damage and that they play a signaling role instead.
In 2015, Hou & Amunugama notice inconsistent empirical results in the area, and try to reconcile them by proposing a model where ROS does cause damage after all. They argue that the antioxidant manipulations (Via gene modification and dietary supplementation), while effective in some cases in flies, failed to work on mice because without external stress, the rate of ROS generation is low enough not to be an issue. But subject to stress, not found in lab conditions, it would be. As for the studies that delete antioxidant-generating genes, they argue that other mechanisms for antioxidant reduction are upregulated, and while infant knockout mice present a higher level of nuclear DNA damage (due to ROS), their rate of increase is slower, so that those mice reach the same level of damage as their wild-type cousins at around the same time, so lifespan is unaffected.
In 2017, Rottenberg & Hoek say that the evidence supporting the mFRTA is extensive, but that in recent years it has been suggested that while ROS may be generated in the mitochondria, they are fine with this, it is oxidative damage to nuclear DNA that is the problem. As for the inconsistent results, the authors note that knocking out SOD1, a gene coding for a key antioxidant protein, does reduce lifespan significantly, but that inducing acute moderate oxidative damage is no problem for the cell, as it can swiftly fix this. Pomatto & Davies (2017) argue the same citing in vitro experiments with oxidant H2O2: A single exposure elicits a compensatory response, but chronic exposure produces lasting damage.
Ren & Zhang (2018) note the existence of the "ROS and aging paradoxes" where there are evidence and theoretical arguments supporting and undermining a role for ROS in aging. They ascribe this to poor measurement and compensatory mechanisms (the antioxidant pathways being more redundant than it is thought).
Barja (2019) pushes back against the role of ROS as signaling molecules and again reasserts that the weight of the evidence is on the side of the oxidative theory.
Finally in a 2019 review, Munro & Pamenter admit as of today no consensus has been reached, and they call for better measurement techniques. The authors even question the idea that mitochondria are even net sources of ROS (!). Where they say that there is consensus is that ROS damage to proteins floating around in the cytosol is not a driver of aging, but damage to the mitochondria may be.
My overall take is that indeed there is no consensus, and that Sinclair's proclamation of the theory as dead was premature. However, a more in depth analysis of the literature would be required to properly weight the evidence. It could well be that a reasonable assessment of the entire body of evidence does point to the theory being refuted, validated, or anything in between. I personally end up thinking that ROS probably plays some role in the aging process, even if it's not the key cause.
Mitochondria
It is almost a meme to say that mitochondria are the powerhouses of the cell, but that's what they really are. They are organelles that float around inside the cell enclosed in a double membrane that houses their own DNA (mitochondrial DNA, mtDNA); them being the only organelles that have this DNA. The regular nuclear DNA has genes that code for the ~1000 proteins that mitochondria need, except for 13 of them or so - the why might have to do with the fact that if the nucleus had those genes, then the proteins would have to be delivered into the mitochondria for the outside and apparently those 13 proteins are not easy to deliver there so they are made in-situ in mitochondrial ribosomes. Mitochondria are constantly being created (By fission of parent mitochondria) and destroyed (If they are damaged, lysosomes break them down); and mitochondria can also merge to form larger units
How do mitochondria generate energy? At a sufficiently high level, by creating an proton gradient between those two membranes. A set of molecular machines, complexes I to V pump the protons into it, which are then subsequently discharged through a rotating molecular turbine, ATP synthase that converts ADP into ATP (A useful molecule that is used for many other processes).
Cells have other ways of generating energy; they are generally fuelled by glucose, which is then broken down to pyruvate, releasing ATP in the process, then that pyruvate goes into the mitochondria where the complexes I-V take the hydrogen atoms (protons) in pyruvate and pump them into the double membrane. During these processes, the mitochondria are using another molecule as helper called nicotinamide adenine dinucleotide (NAD) which exists in two forms: NAD+ and NADH (The latter is the former plus an extra proton).
There have been theories proposed to explain part of the aging process resorting to ROS and mitochondria as noted in the earlier section, but it is worth mentioning the theory proposed by Aubrey de Grey - of SENS fame - which is not referenced in the reviews I have linked earlier but that seems to explain a bunch of the inconsistencies the research founds. In particular, the facts de Grey (2005) and (2002) starts from are:
- Mitochondria generate ROS
- Not much ROS does escape mitochondria (And thus they don't affect proteins in the cell)
- Because of the above, an increase of antioxidants in the cell does nothing to affect ROS or reduce damage
- ROS damages mtDNA (And this is why evolution has been moving almost every gene from the mitochondria to the cellular nucleus, save for the 13 genes that are hard to migrate)
- Deficiency in SOD2 (An antioxidant enzyme that works inside mitochondria) reduces lifespan in mice
- Cells either contain healthy mitochondria, or (In a tiny minority of cells), mitochondria that all have the same mutation (in genes that code for some of the essential proteins for complexes I-V or ATP synthase) and are inactive.
His reasoning is then that while most damage to mtDNA leads to damaged mitochondria that are garbage-collected and replaced by new ones; if a mutation stops ATP production and thus ROS generation, the mitochondria won't experience damage and will stick around; eventually, as other healthy mitochondria are replaced due to damage, given than the defective ones are never garbage-collected (As they are not damaged), those end up dominating the entire cell. Once this happens, this disrupts the balance of NAD+ to NADH which then the cell tries to balance out in a process that ends up exporting ROS outside the cell; this ROS then reacts with -among others- LDL chosterol, oxidising it, and then this damaged form of cholesterol can cause damage to cells elsewhere.
If damage to mtDNA is a cause of aging, a way to avoid it is to introduce copies of the remaining 13 genes in the celular nucleus. That way, regardless of what happens to mtDNA, there is always a way to make the relevant proteins. This is known as allotopic expression, and there has been success so far in moving 4 of those 13 genes in vitro (Made Artika, 2019)
Recently, it has been found that mitochondria can also be found circulating in the blood. It's still unknown how they get there or if they play any function; likewise a recent finding is that cells can share mitochondria with each other. This is all to say that biology is incredibly complex and we are still discovering basic facts about it.
Cellular senescence
I've mentioned senescence earlier a couple of times. If a cell is damaged beyond repair, it can either die (apoptosis) and be recycled, or stick around and become a senescent cell. Senescent cells have a Senescence-Associated Secretory Phenotype (SASP) that in acute doses signals to the immune system that those cells should be cleared. But if there are too many senescent cells, then the SASP has truly negative consequences for the cells around them: A component of the SASP is the emission of cytokines and chemokines (proteins with signalling functions that regulate the activity of the immune system) to the extracellular medium that cause inflammation in neighbouring cells.
In aged individuals, the % of senescent cells is usually <20% of the total number (Yang & Sen, 2018) , although this varies by tissue (Tuttle et al. 2019). In mice, young individuals have 8% of senescent cells, compared to 17% in old age (Lopez-Otin et al. 2018)
To complicate things, senescent cells are not bad in general, they also help during embryonic development, tissue repair and wound healing, and of course the mechanism that turns cells senescent to begin with is in place to prevent cancer (Victorelli & Passos, 2017). On that last point, it also has to be said that senescent cells, through inflammation of nearby cells, promote cancer (Lee & Schmitt, 2019, Prieto & Baker, 2019); and eventually a senescent cell can escape that state of arrest and turn cancerous too.
A commonality in senescent cells is the expression of the protein p16INKa which helps to identify them in vitro; however externally senescent cells are hard to distinguish from regular cells. A very recent review of it, a "SASP atlas" is Basisty et al. (2020), shows that there may even be different kinds of senescent cells, some of which have the positive effect noted earlier, and others just abnormal, negative effects.
Their effects are so negative that even transplanting a small amount of senescent cells to other mice causes them to live less and become less healthy (Xu et al., 2018)
Autophagy
A mechanism cells have to fix damaged component is simply to break them apart, and produce new ones from scratch: autophagy (Bareja et al., 2019). It is at least intuitive to think that throwing something and building it again from scratch is easier than trying to repair a broken component, so it's not that surprising that autophagy is involved in the alleviation of most if not all of the hallmarks of aging - to be discussed in the next section-.
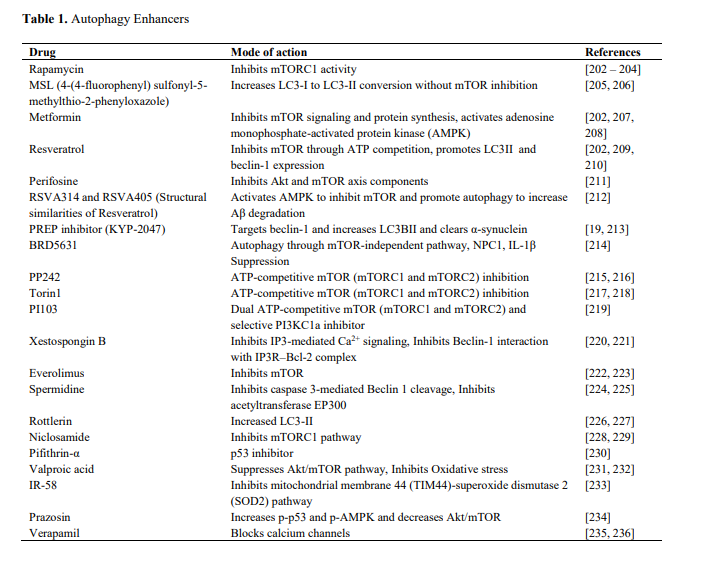
Interventions that target the various longevity pathways (AMPK, mTOR, IIS, sirtuins) also tend to upregulate autophagy. Here's a list of drugs that promote autophagy (From Shetty et al., 2018)
The role of the brain
The nervous system plays a role too. Zhang et al. (2017) found that the hypothalamus' neural stem cells (NSC) secretes small pieces of RNA in vesicles (exosomal miRNA), and their rate of release decreases with age.
Note that for a while it was thought that there were no stem cells in the brain and thus no renewal of neurons. Now we know that in some select areas of the brain there are some, so far they have been found in the hypothalamus both in mice and humans (Pellegrino et al., 2018)
Using a virus to remove a few of these NSCs, or implanting NSC into mice the lab showed a decrease in lifespan after removing these cells (In mice 250 days old), and conversely an increase when implanting them (In mice 600 days old), in this latter case extending maximum lifespan by around 13%.
It has also been found that the genes coding for certain proteins (REST) related to neural activation are more expressed in brains of long lived individuals. Genetically engineered worms with lower activity of REST live shorter lives, and in mice increased REST seems to activate FOXO1, a transcription factor that has elsewhere been found to delay aging.
Tricking mice into thinking that they are colder than they are also has positive effects (Conti et al., 2006): raising the temperature of the hypothalamus tricks it into thinking the whole body is warmer, and the hypothalamus tries to compensate by, seemingly, making cells more efficient and thus generating less heat. This increases median lifespan by 12-20% (male/female) in mice, but does not extend maximum lifespan.
Inflammation
When the body detects pathogens, or otherwise anything that shouldn't be there, specialised cells released small peptides called cytokines which attract immune cells (macrophages, lymphocites, etc) to the site to deal with the pathogen. There are various signs associated with it including pain, a burning sensation, redness and swelling; the root cause being vasodilatation to facilitate the arrival of immune cells to the area.
This all sounds good and reasonable, but what does it have to do with aging?
Remember the senescent cells? Part of the SASP is the emission of cytokines, a way of telling the immune system to clear them out. But if they are not (Because there are too many, or because the immune system is weakened) then the result is chronic inflammation around the whole body. Other factors also lead to this age-related kind of inflammation, which is typically referred to as inflammaging in the literature (Franceschi et al., 2018, Xia et al. (2016)). A related concept, metaflammation refers to inflammation for metabolic reasons due to overeating, a reason that helps explain why calorie restriction works: eating triggers an inflammatory response to prepare the body against potential pathogens in the newly ingested nutrients.
Inflammation is a response to a perceived threat that helps fight it, but inflammation even in the absence of any pathogen is perjudicial for the body; else we would have evolved to enjoy inflammation all the time. Inflammation swells tissues, which cause nearby tissues to expand and tear apart at the cellular level; the constant tearing up and rebuilding can lead to fibrosis (Mack, 2018). Also, the environment that inflammation creates is one that is meant to increase cell turnover (More apoptosis, but also more cell growth to replace lost cells), with granulocytes secreting toxic agents (Including ROS) to make the area affected less hospitable (But also increases damage to DNA), and specific cytokines like the tumor necrosis factor that induce cell death, and growth factors that promote cell growth. This increased turnover can facilitate the appearence of cancer.
Aging
As times goes by, the probability that a given organism dies increases. In humans, this can be modelled by the Gompertz-Makeham (Not quite, at birth there is a higher change of death, and in later age some people have argued that mortality may taper off, staying constant.) law of mortality that relates an age to the probability of dying on any given year. The law has two components, one is a constant that is independent of age (Think accidents or infectious disease) and another is age dependent and increases exponentially. In an animal that does not age, the probability of dying is the same every year. In good environmental conditions, this would be negligible. Here's what the empirical data shows for humans:

Some species age very slowly, or don't age at all, and this is not limited to small or simple lifeforms: The Greenland shark has been observed to live over 3 centuries. The fact that these amazing lifespans have been observed in the wild give us some hope that one day we'll copy the mechanisms they use to live to such long lives and reproduce them in humans.
The relevance of aging as a problem is something I won't discuss at length here, but suffice to say that inasmuch as longer -and healthier- lives get us to experience more of what we like, curing aging magnifies the impact of all other interventions that positively benefit us.
Why we age
It is easier to see how aging works by looking first at how things usually work. So we have DNA being made into proteins, everything is working the way it should. Damage occurs for various reasons, it can be ROS, it can be random mutations in DNA, DNA breaks, or just molecules bumping against each other in the wrong way. The cell has repair mechanisms to handle all of this, and they mostly work. The genome itself is quite robust, and even when mutations occur, they are mostly harmless. But the epigenome is more brittle: The same flexibility it needs to be able to be switched on and off depending on the tissue and the situation ends up being a problem. With the loss of epigenetic information, the rate of errors the cell makes during its function increases: everything that could go wrong - from misfolded proteins to an accumulation of garbage (lipofuscin) due to insufficient clearing - goes wrong. This eventually leads up to failure, be it in senescence, cancer, or apoptosis. These failures interact in nasty ways: The weakening of the immune system leads to impaired clearance of senescent cells. And at the same time more cells are turning senescent due to shortened and damaged telomeres. Senescent cells induce nearby cells to overexpress CD38, consuming NAD and disrupting sirtuin activity (Chini et al., 2019). Maybe even telomere shortening represses sirtuins too (Amano & Sahin, 2019)
There is disagreement as to why this may be, there being two explanations. One, the majority view, is that selective pressures are much weakened after the reproductive age, and so it would have been hard for evolution to steer our genomes towards living longer if that doesn't lead to more offspring. Additionally, deleterious mutations in DNA that only become a nuisance later on in life could accumulate, as those wouldn't reduce fitness in the individuals capable of reproduction earlier in life. And finally, some mutations that are explicitly selected for their usefulness early in life can be harmful later on (antagonistic pleiotropy).
Second, a minority view is that aging is programmed, that those mutations that cause aging are there because in the aggregate make the species fitter, even when they make a given individual less fit. See Kowald & Kirkwood, 2016 for a critique of the programmed aging theory, Barja (2019) for a defence.
Many theories have been proposed in the past that point at correlates of aging like DNA damage or ROS, but now evidence seems to indicate that most of these are downstream of the root cause of aging. A review of some of these historical theories is Kunlin 2010 or Kochman 2015 .
The Hallmarks of aging
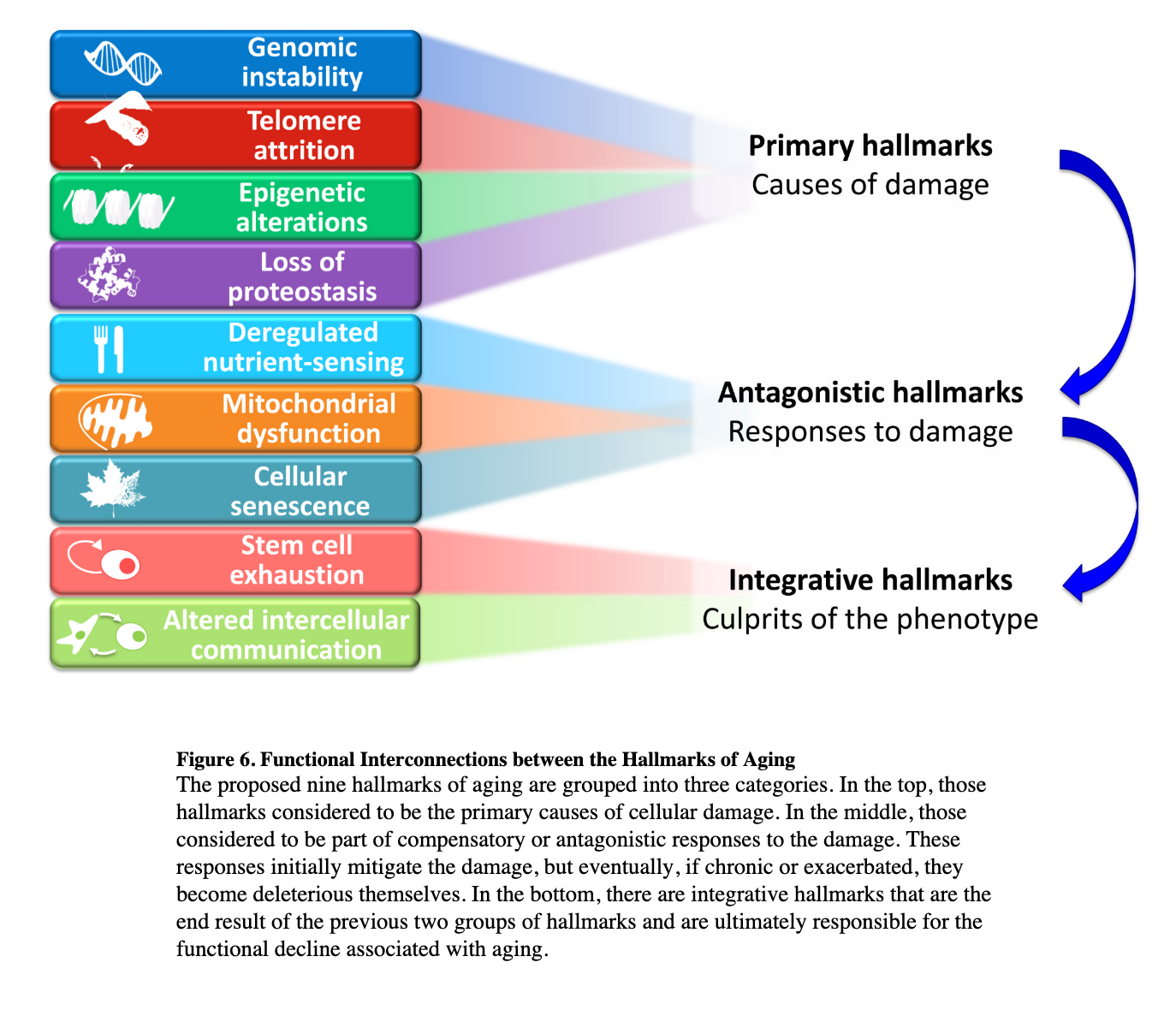
Aging of the human body is aging of the cells that compose it. When we think of the telltale signs of aging, we think of things like wrinkles, loss of cognitive function, lower bone density, impaired motor skills, whitening of the hair, or loss of vision. One can try to find common factors in the cells involved across our organs to see what an aged cell looks like. In doing so, we encounter the hallmarks of aging (Lopez-Otin, Blasco et al., 2013, Dodig, Cepelec and Pavic, 2019, Ferrucci et al. 2019). I'll discuss here the four core or root hallmarks, the rest can be found in a summary diagram below, and in the paper itself.
Genomic instability
The DNA gets degraded both in structure and in information encoded due to exposure to reactive chemical species, replication errors, or other factors. This is also true, perhaps even more true, for the DNA of the mitochondria (mtDNA), as mitochondria are the locus of chemical reactions that deal with highly energetic reactions, the subproducts of which can over time damage the mitochondria itself. The Lopez-Otin paper mentions that in more recent times, it is thought that the issue with mitochondria is more due to replication errors, and not so much due to oxidative stress, a result that later was found to be associated with early stage Alzheimer's (Hoekstra et al., 2016).
Telomere attrition
As explained earlier, telomeres are the end "caps" of chromosomes, and they get shorter with each cell division. When they shrink too much, the cell can't divide anymore (The so-called Hayflick limit). An enzyme, telomerase, is capable of extending them, but most cells in mammals have the genes required to produce telomerase switched off. If forced to express it, cells can effectively live forever. But even with telomerase, telomeres themselves can become damaged, and due to the presence of the protein complex shelterin in the telomeres, the enzymes responsible for repairing DNA do not act on the telomeres. This telomere damage can lead to senescence and/or apoptosis (cell suicide): If the telomeres are so short shelterin can't even bind to it, a signal is sent that triggers senescence or cell death (Schmutz and de Lange, 2016). One may think that perhaps getting rid of shelterin would be a good idea as this would allow the telomeres to be repaired too. But this has been shown to lead to death even at the embryonic stage: The cells panics if there is no shelterin in the telomeres. As discussed in the earlier section about telomeres, their status as biomarkers of aging has mixed support, and their role in aging is probably through damage sensors that in turn drive cell senescence, and this in turn generates further damage in combination with a weakened immune system, unable to clear the senescent cells.
Epigenetic alterations
We discussed the epigenome and epigenetics earlier. Here, the carefully arranged set of DNA and histone methylations and chromatin coiling becomes disrupted with age, leading to irregular levels of gene activity. A pattern that seems associated with aging is a generalised loss of methylation. Most of the DNA is methylated, and methylations suppress genes that are not needed for the function of that cell. But also some locations become hypermethylated, including those of oncogenes, The authors of the Hallmarks paper (published in 2013) note that thus far there is no direct experimental demonstration that organismal lifespan can be extended by altering patterns of DNA methylation. (The case is true for histone modifications, where most experimental work has focused).
As I will explain later, damage to the epigenome is in principle reversible - this is not the case, or at least not trivially, for DNA mutations - .
Loss of proteostasis
Protein homeostasis is the state of a cell when the concentrations of proteins is the right one for cell function, and when deviations from it can be returned to equilibrium by the cell through various mechanisms such as proteins that help other proteins fold themselves properly (chaperones like heat shock proteins ), or degradation of incorrectly folded proteins in lysosomes or proteasomes:
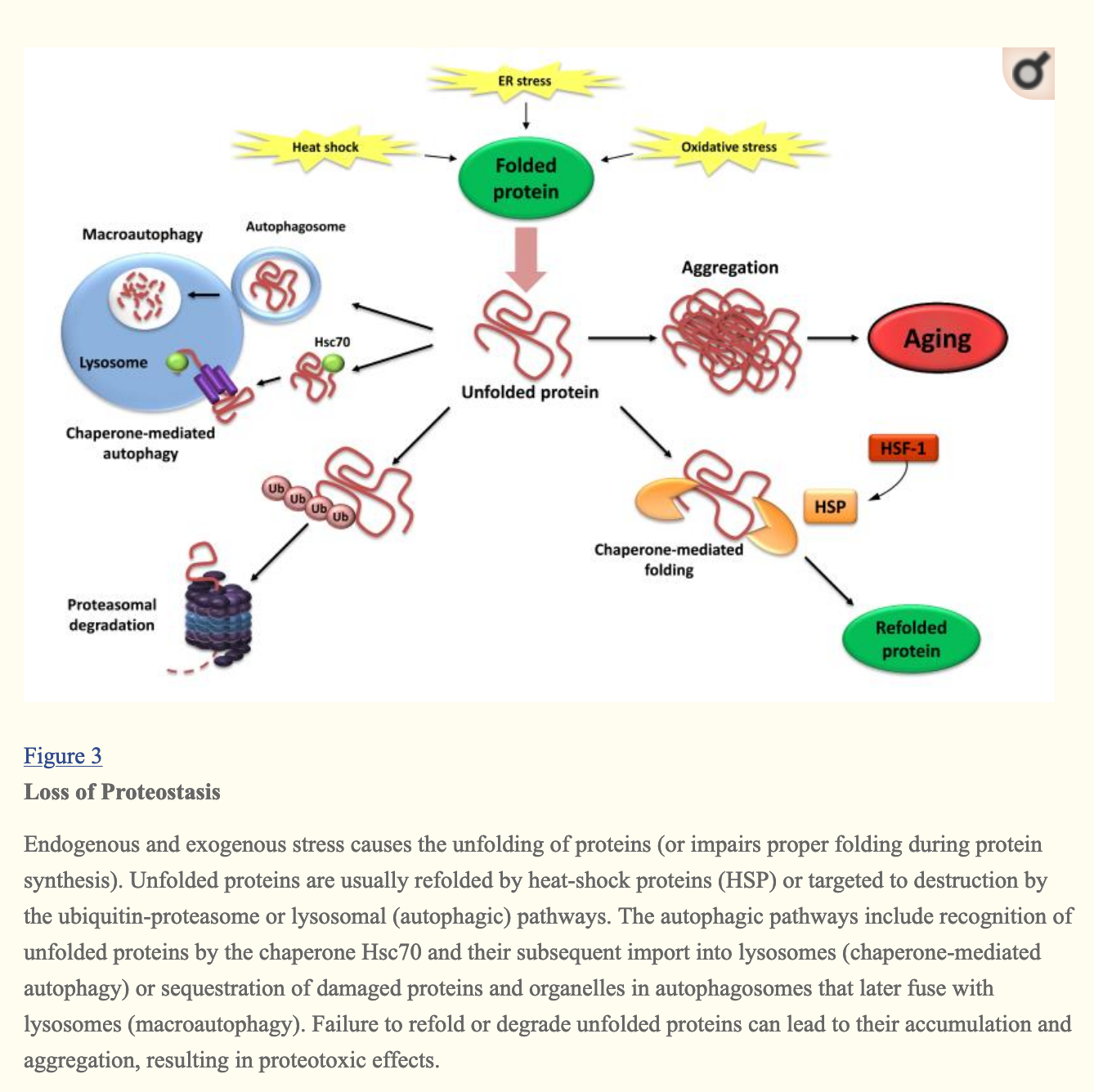
The SENS typology of damage
Prior to the Hallmarks, SENS CSO Aubrey de Grey proposed the existence of seven kinds of damage, the goal being to devise a classification that is exhaustive (Covers all possible damage) and divides the damage in such a way that can be addressed by one kind of therapy by category. This is how the SENS categories relate to the Hallmarks:
| Hallmark | SENS damage kind |
|---|---|
| Genomic instability | Cancerous cells |
| Telomere attrition | Death-resistance cells, cell loss (Indirectly) |
| Epigenetic alterations | ? |
| Loss of proteostasis | Extra & Intracellular aggregate formation, extracellular matrix stiffening |
| Deregulated nutrient sensing | ? |
| Cellular senescence | Death-resistance cells (senescent cells) |
| Stem cell exhaustion | Cell loss |
| Altered intercellular communication | Death-resistent cells (SASP) |
| Mitochondrial dysfunction | Mitochondrial mutations |
History of aging research
AGE-1 and the insulin pathway
It all began with a worm (Johnson, 2013, Kenyon, 2011), Caenorhabditis elegans. In the 80s, it was known that longevity was heritable in yeast, worms, and flies, and so finding genes that mediated this process seemed like an obvious thing to do. Michael Klass, a postdoctoral researcher at the University of Colorado was trying to find mutations in worms that would lead to longer lifespan; this was achieved by exposing worms to EMS, a mutagen. He found that caloric restriction was also known to be effective to prolong lifespan in worms, but it was not known why that would be the case. Some mutations were leading to longer lifespans, but because they seemed to restrict feeding, causing caloric restriction.
Thomas Johnson, another postdoc at the same university, eventually showed in 1988 that there was a gene, age-1 (Or, because other people discovered the same gene but initially didn't know it was the same gene, it also got the name daf-23) in C. elegans certain mutations of which prolonged lifespan up to 70% without inducing caloric restriction, but with reduced fertility. Later on he would show that it's possible to retain these effects without the reduced fertility. An interesting fact from this story is that one of the now leading researchers in the field, Cynthia Kenyon, deliberately decided not to work on the further study of age-1 because "in our friendly C. elegans culture, it was not polite to study someone else's gene, and age-1 belonged to Tom. But mainly, I wanted to carry out my own screen for long-lived mutants, to see what came out."
One early hypothesis for age-1 was to see if the mechanism by which it prolonged lifespan was by diverting resources into cell maintenance that would have otherwise been used for reproduction. At her lab, Ramon Tabtiang, a graduate student used a laser to remove the cells in young worms that eventually develop into their reproductive organs, though one may argue that perhaps there was something in the intervention itself that harmed the worms. This made the worms sterile, but did not lead to increased lifespan, against the prediction of the resource diversion theory. Kenyon also found that daf-2 mutant worms had an even greater increase in lifespan, up to 100%, without loss of fertility or any other negative consequence. Years later, the finding would be confirmed using more precise techniques (Rather than mutating the worms and praying that the relevant gene is mutated, using RNAi to knock down the activity of the gene in a more precise way). Furthermore, she found that another gene, daf-16 (aka FOXO3 in humans) intervened in the process leading to longer lifespan. In adult worms, reducing the expression of the daf-2 gene prevents daf-2 from reducing the action of the daf-16 gene, in turn increasing longevity.
To make things more interesting, if you get rid of - just the gonads, instead of the sac that holds them entirely -, you again get a boost to lifespan! (Hsin and Kenyon, 1999)
What does daf-2 exactly do? It codes for a receptor (This is, a protein that sits in the cellular membrane that senses molecules outside of the cell) that is an ortholog (A genetic equivalent) of the IGF-1R receptor in humans. This receptor in humans detects the Insulin-like Growth Factor (IGF) which is similar to insulin (C. elegans doesn't have insulin; they have insulin-like peptides). As you can see from the picture below, activation of the receptor (daf-2 in the picture), through the activation of various intermediate proteins eventually inhibits the transcription of daf-16/FOXO. In turn, the FOXO protein regulates multiple other genes that affect longevity. To sum up: ↓ daf-2 → ↑ daf-16 → ↑ activity in longevity-promoting genes → ↑ longevity.
In turn, what does this have to do with caloric restriction? Lack of nutrients means less activation of the IGF1 receptor encoded by daf-2. If so, then, if this was the only pathway involved in sensing nutrients, we should expect that for the worms that already had reduced activity of the receptor (for example due to a genetic manipulation), caloric restriction won't increase lifespan at all (Not quite, as there are other pathways; daf-2 mutants subject to CR live longer, Lee et al. 2006, Zhang & Mair, 2016).
Is the same case for the gene that Johnson found, age-1/daf-23? A laboratory technician, Jenny Dorman found that this is also the case: Without daf-16 activity, decreased activity in age-1/daf-23 no longer leads to longer lifespan.
In red below is the pathway we've been discussing, plus a few other proteins in the same pathway that were discovered later (PDK1, AKT, and daf-18).
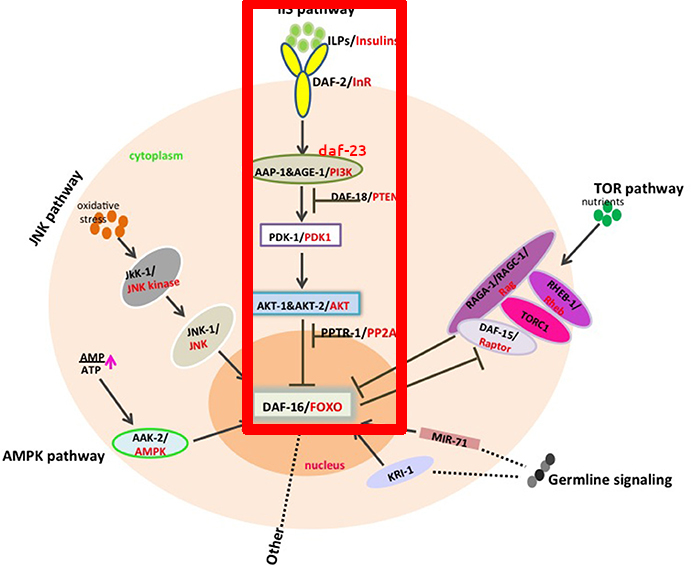
(Figure from das Neves, 2017, highlighting+ daf-23 mark is mine)
The AMPK pathway and TOR
Starting in 2000, some other pathways have been added to the two above: AMPK (AMP kinase) is upregulated when energy is limited, and the uncreatively named TOR (Target Of Rapamycin, or mTOR in mammals) is a nutrient sensor that, when downregulated -by rapamycin - also promotes longevity.
Downregulation of TOR causes more autophagy and in particular mitophagy, whereby mitochondria are recycled, and upregulation in AMPK (along with sirtuins) helps prevent oxidative damage in mitochondria.
Guarente is unsure how many of the hallmarks of aging can actually be influenced by the pathways described here, or how many negative effects there will be in humans if one intervenes in them. For the case of rapamycin, apparently its effects are uneven by tissue, in some cases even causing damage (in mice), excessively blocking the IGF1 receptor (The one encoded in worms by daf-2) in humans could lead to poor cardiac health and loss of muscle mass. Activation of SIRT1 could help certain tumors grow in some contexts in conjunction with the expression of oncogenes (Like n-myc) or when tumor suppressor HIC1 is downregulated, even though it normally has a tumor-suppressing effect (Stunkel & Campbell, 2011).
You can find more in Zainabadi (2018).
The genetics of longevity
As with everything else, longevity is heritable, but not as much as one would expect, just around 10-30% in the most recent estimates,depending what you control for (Graham Ruby et al. 2018). As for looking at genes directly, of course the variance explained is lower ("missing heritability"), at around 8% (Wright et al., 2019). If you are curious about your own case, you can upload your sequenced genome data to Promethease, search for 'longevity' and it will search and sort results from SNPedia for you, or you can manually have a look here. The effects seem incredibly large, e,g having the right allele of rs2802292 will up to triple your odds to live to 100 years, rs2802288 increases odds of living to 100 by 1.5x, at least in the populations tested.
Thus one option to lengthen lifespan is to genetically engineer embryos to have the beneficial alleles of these SNPs (SNP being a position in the genome where there are at least two variants in more than 1% of the population. We are 99.9% genetically equal, so SNPs is a way to look at those genes that makes us different)
Interpreting longevity research
Animal models
Research on the mechanisms that drive aging and potential therapies to slow or reverse it is usually done on a series of animal models: yeast, flies, worms, and mice. As mentioned above, the pathways involved in aging are more or less preserved across species, but there are substantial differences that one has to be aware of that can cause a treatment not to work in humans.
Take telomerase. It's not active in most human cells, but in mice it is (Shay & Wright, 2019). Or cell replication. Cells replicate in humans (Well not always, heart cells do not), and in any given point we have different numbers of cells. But not in the worm C. Elegans, which has around 1000 cells and that's it. Mice tend to be overweight, which can confound the effects of some interventions like calorie restriction. In general, non-vertebrates do not present senescence, stem cell exhaustion, immunosenescence, or inflammation. (Singh et al. 2019).
There is a paper, Liao et al. (2010) that claims that CR has negative effects in some strains. However upon closer examination this doesn't seem robust: There were 5 mice per strain, and some of the more extreme negative results recorded involved just 2 or 3 mice (because they couldn't get enough). In contrast, I couldn't find any papers with a larger sample size showing negative effects of CR in mice in lifespan. To be sure some papers show neutral effects (no increase in lifespan). There are some cases, like in DBA mice where CR of 40% started early in life increases early mortality. The net effects seem neutral (eary mortality+lifespan increase for the individuals that survive). 40% CR is quite extreme; people that do chronic CR rarely go this far. More recently, Unnikrishnan et al. (2021) repeated Liao's analysis with a larger sample (though, perhaps, not large enough!) with the expected results: Liao had found a worst case scenario of the 115-RI mouse line where lifespan went down by 70%, Unnikrishnan et al.'s sample increased their lifespan instead as the model would predict, though this effect was markedly stronger in females than males. Interactions between genetic background and the effects of CR are real, but the reasons to be pessimistic are not as strong as one may infer from Liao's paper.
Here's a summary of pros and cons of using diverse model animals. One example not there is daphnia .
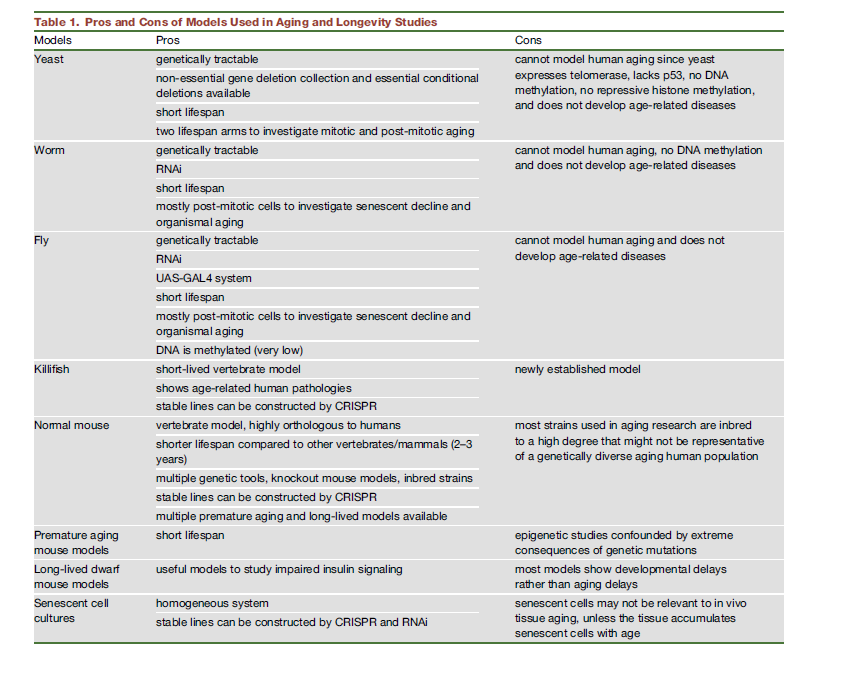
Note that this may be already out of date! DNA methylation may occur after all in C. Elegans!
One more animal that is now being looked at is the Naked Mole Rat (NMR), as it doesn't seem to age. NMRs in this regard show elongating or constant size telomeres with age, while Spalax (a cousin of the NMR that is also long lived) shows shortening of telomeres (Adwan Shekhidem et al. 2019). The NMR is so interesting that it deserves its own section, but I won't have space in this FAQ for that.
Measurement and experimental issues
Unlike in other disciplines where measurands are crisply defined and can be measured with extreme accuracy, in biology it is not the case. This for example has been the source of inconsistencies in telomere research (Sanders et al. 2013, Simons, 2015), and the use of quantitative PCR (qPCR) as a common assay has meant that researchers had a hard time looking at what mattered (The shortest telomere). More modern assays like TeSLA or FlowFISH have been developed that have lower variability. (Shay & Wright, 2019). Martin-Ruiz et al. (2014) illustrates this by sending 10 same samples to 10 labs each and having them measure telomere length with various methods, finding substantial variation.
Some reviews warn that studies should always look at both health and lifespan, complaining that in the 20 years prior this has not been done as much as they'd like (Bansal et al. 2015), reporting that some mutations in worms that prolong lifespan do not always lead to extended healthspan, so the worms stick around for longer but in a decrepit, zombielike state. Researchers from the Kenyon lab replied to this work (Podshivalova et al., 2017), and acknowledged that this is the case for the particular mutations studied, but this is due to enhanced bacterial resistance in the mutants, so if both sets (wild-type and mutants) of worms are fed dead rather than live bacteria, both wild-type and mutants live longer, and present a decrepit end-stage, but comparatively the mutants have more healthy days in their life.
Regarding the methods themselves, it's worth noting that knocking out genes may result in other unexpected changes in function, unrelated to the main function of a given gene (He, 2016).
It used to be believed and some papers still claim this (see Wagner 2019, reviewed by Steve Horvath himself, or Sen et al., 2016), that the general pattern that occurs with aging is generalised hypomethylation, coupled with local hypermethylation at some sites. The data indeed showed this, but this was due to subpar measurement techniques (Unnikrishanan et al., 2018).
What this kind of research looks like
The way the papers are usually laid out is first some small molecule, protein, gene change, etc that is to be trialled that is applied to a treatment group. There tend to be assays to confirm exactly what is going on, but ultimately if we are looking at individual deaths, we can take note of how many individuals were alive at the beginning and how many die at each point and plot this. Here's one such example from that 1999 Hsin-Kenyon paper that shows wild-type C. elegans worms with various treatments (Here, the regular worm is the black square, and the others have various ablations to their gonads or germline cells). From these plots, one can look at the age when 50% are dead: This is the median lifespan in that treatment. Or one can look at the age of the oldest individual when they died, this is the maximum lifespan. In some papers, it is sometimes not clear what they are talking about (Median or maximum).
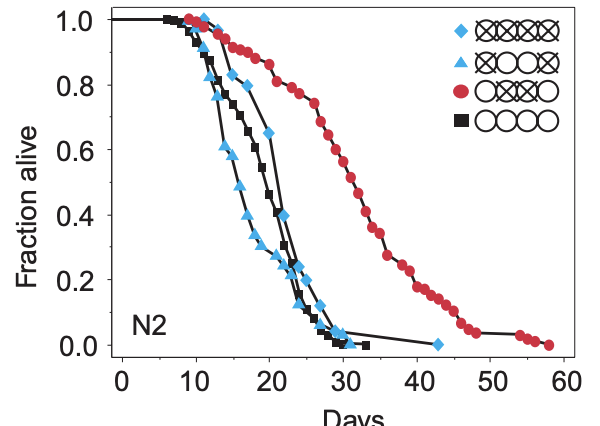
The way to know that an intervention does what it does for the reason it is thought that it does is applying some form of gene editing, generally a knockout (Removal of a gene) or knock down (Reducing its activity). For example, if we say that resveratrol enhances longevity, and this is done via the activation of sirtuin SIRT1, then if we knockout SIRT1, resveratrol should have no effect.
Interventions
Longevity research sometimes resembles nutrition research: study quality is not very high and there are many things that could go wrong or make an experiment different from another to make extrapolations difficult.
Fortunately, the National Institute for Aging has the NIA Interventions Testing Program, or ITP (Nadon et al., 2017) where they test the same thing in three labs with larger sample sizes and rigid experimental protocols. Their stage one uses around 100 mice per group, while their more rigorous second stage tests use up to 500 mice across all the labs per group. This testing has revealed as of 2017 six compounds that extend life in mice. Some of them only worked on males, others on both male and female mice, and in those the effect by gender were different. Another review I'll be using for this section is chapter 19 from the handbook of models for human aging ( Unnikrishnan et al., 2018 )
| Drug | Median or mean lifespan | Max lifespan |
|---|---|---|
| Aspirin (Only males) | 8% | ~0 |
| Rapamycin (females >males) | 23% (m) / 26% (f) | 8% (m) / 11% (f) |
| 17αEstradiol (Only males) | 12% | ~0 |
| Acarbose (Males >> females) | 22% (m) / 5% (f) | 11% (m) / 9% (f) |
| Nordihydroguaiaretic acid (Males only) | 12% | ~0 |
| Protandim® (Milk thistle, bacopa, ashwagandha, green tea, and turmeric) (Males only) | 7% | ~0 |
What did not work? Metformin, fish oil, ursodeoxycholic acid, resveratrol, green tea, curcumin, or the MitoQ supplement, among others. You can find everything they have tested, or that is being tested here
Note that the fact that these have not worked in mice does not imply that they will not work in humans. Some things work in mice and not in humans, and viceversa. An example may be fish oil; while some recent evidence seemed to show that supplementing omega 3 did not reduce cardiovascular disease or mortality in general, a later meta-analysis (Hu et al., 2019) claims the opposite with a much larger sample size, although the effects seem very small.
Another thing to note here is what counts and does not count as an anti-aging intervention. Antibiotics or vaccines have increased life expectancy, but not by slowing down aging. The fact that they are not discussed here doesn't mean they are not effective to add years of life. The baseline against which I compare these interventions is a healthy (non-obese, non-smoker) human individual doing moderate exercise, eating a whole foods plant-based diet, a Mediterranean diet or something along those lines. For that individual, what can be done to make them live longer?
A quick summary of some drugs that some people believe might help in humans is here, broken down by key target (From Pan and Finkel, 2017):

Gwern did a preliminary cost-benefit analysis of vitamin D and metformin; in terms of decision-making one should weigh the evidence and on the basis of that decide what to do, and this is what Gwern does there; even if the evidence is weak, if a treatment is cheap and there are not side effects, a slight chance of it actually working justifies taking it.
Calorie restriction / Intermittent fasting
This is the most well known and studied intervention to extend life across species. The pathways that are involved with CR are the same ones that I mentioned earlier, and so due to the precedence of CR in the hierarchy of longevity interventions, these other drugs sometimes receive the name of CR mimetics.
CR interventions typically involve cutting food consumption 30/50% with respect to what the animal would eat if food is always available (ad libitum feeding). Some authors use the term Calorie (or caloric) restriction to refer to reducing calories, but keeping nutrients like vitamins constant, while others use dietary restriction (DR), where no supplementation is included, so these animals receive less calories and micronutrients.
Another intriguing result here is that of selectively reducing the availability of certain nutrients, in particular the essential amino acid (That is, an amino acid an organism cannot make on its own), methionine. The handbook reports a range of 7-30% for median lifespan increase and 9-11% for maximum lifespan.
The literature on calorie restriction in general has been criticised for comparing ad libitum fed animals with CR. Ad lib mice tend to become obese, and so perhaps it is obesity that's bad, rather than CR having a positive effect (Sohal & Forster, 2014). The authors go as far as claiming that as of the publication of their study, CR has not been shown to extend species-maximum lifespan (wrt healthy individuals)
As to the how CR would work, it has been hypothesised that through mTOR, as inhibiting proteins downstream of the mTOR pathway negate the longevity effects from CR, as well as through the IIS pathway, as ultimately CR is affecting the levels of circulating glucose and insulin, and also via the activation of sirtuins.
The best evidence to see how this would translate to humans are some studies done in macaques by Mattison (2013) and Colman (2014) and jointly reviewed by Mattison, Coleman, et al. (2017) These two studies are interesting, because macaques are closer to humans than rats or worms, and so the effects that we would expect in humans should be more similar to what we see in macaques. Some recent analysis (Vaughan et al., 2018 , Le Bourg, 2017) claimed that given the results in macaques, it seems we have evidence that CR does not work in humans, but there is more to it. Here's the key differences between both studies:
| Mattison (NIA) | Colman (UW) | |
|---|---|---|
| Animal | Indian & Chinese macaque | Indian |
| Sample size (Control/CR) | 64/57 | 38/38 |
| Diet | Natural (Whole grains, soybean meal, etc), soy, corn, fish oil. Higher in protein and fiber wrt UW. | Semi-purified (Made from soy protein or sugar, carefully formulated), corn oil |
| % sucrose in total carbs | <7% | 45% |
| Feeding | 2 meals per day | 1 meal per day |
| Age of onset of CR | Young, adult, old | Adults |
| Median lifespan change | ~0 | ~9% |
| Average calories per day in control group (17 y.o., males) | 720 | 840 |
| CR wrt control (males) | -26% | -24% |
| Healthspan increase reported | Yes (But p=0.06) | Yes |
In neither of the studies the monkeys were fed ad libitum; instead an appropriate amount of food was calculated beforehand based on age and bodyweight, this baseline was based on published standards (in the NIA study) or on an individual per monkey baseline based on their consumption prior to CR (in the UW study), the reasoning of this being that this is closer to how it would work in humans. Additionally in the UW study, food was adjusted through the experiment for the control group so that at the end of the day there was always leftover food, ensuring that monkeys were eating as much as they wanted.
In both studies, monkeys that died due to conditions that are not age related were not considered for survival calculations, but the policies to treat conditions were different: In the UW study, any medical condition was treated, in the NIA study only those that caused suffering were treated until 2010 when they started treating at least endometriosis, as many female monkeys were dying from it.
Looking at the evolution of the weights across the studies may be misleading because of the different species used, but adiposity (grams of fat per gram of total weight), being a ratio is less biased, here in the NIA study there is almost no gap between CR and control, while in the UW study, the control monkeys are visibly more fat; but not only that: Taken in the aggregate, the NIA control monkeys are indistinguishable from the CR UW monkeys (in adiposity). This is most likely from that excessive sucrose consumption. It has to be noted that in the NIA study there was greater genetic diversity (As they were sourced from different origins) so it could be that this made it more difficult to precisely calibrate the right dose of CR, reducing the observed effect.
Looking at health, monkeys were assessed on the age at which developed certain age related diseases (including sarcopenia, osteoporosis, arthritis, diverticulitis, cataracts, heart murmurs, cancer, diabetes, or cardiovascular diseases)
In the UW study, the probability of dying on any given period was estimated to be almost twice for the control group compared to CR and the likewise for health conditions this was 3x in the control group. In the NIA study, the probability of dying was not different, but the control group was twice as likely to develop some of the above condition by some given age. While this difference was reported as not statistically significant in the NIA study, looking at the survival plot below it is very plausible that CR helps; however it seems to have significantly decreased the incidence of cancer and diabetes.
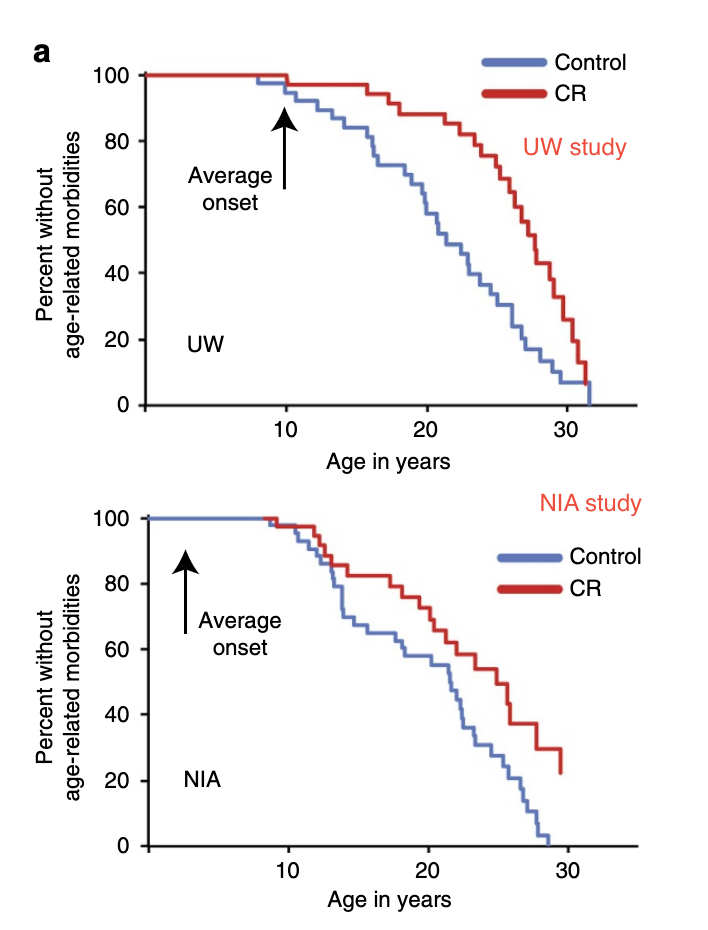
The authors claim the reason the NIA study found a null result was that their control group de facto was calorically restricted and conclude that further caloric restriction after a given point does not further help.
On the basis of the evidence provided above, I conclude that caloric restriction in monkeys improves health by some amount, and reduces cancer incidence and diabetes. If it also extends lifespan, it does so by a small amount of less than 10%. This is of course conditional on the baseline of a reasonable diet, it could be argued that in the human case, we effectively eat when and what we want, so we may be like the control group in the UW study rather than the NIA one. In any case, the benefits we would be likely to see in humans are not the 80% increases in lifespan seen in some rat studies, as is generally the case that long-lived species see smaller effects from a given intervention.
If so, then we should be more optimistic, and indeed taking regular people and subjecting them to 10% CR (They were told to do 25%), as it was done in the CALERIE trial improves biomarkers of cardiovascular health (Kraus et al., 2019). There are also other, less clear pieces of evidence (natural experiments, correlational studies from populations who tend to eat less as in Okinawa, and members of the Calorie Restriction Society) that also point in the same direction (Most et al. 2017). The same paper also warns that, as one might expect, severe CR is not good for you.
Thus, calorie restriction, given that it's cheap (In fact, it saves money) and saves time (In cooking) and has some health benefits, I would recommend everyone to practice it unless there is an overriding reason not to (Like some medical condition). But perhaps it is possible to get the benefits of CR without feeling chronically starved: intermittent fasting.
In an adversarial collaboration in the Slate Star Codex blog, the authors conclude that indeed CR does increase lifespan, but warn against adopting it due to negative effects on fertility or increases severity of infections, plus psychological changes (Obsession with food, lower motivation). While some of these negatives are disputable (Lower fertility and libido are okay if one does not desire to procreate at the time), I think the rest will probably not occur with a CR, or especially under intermittent fasting, described below. The authors also didn't include the CALERIE trial due to adherence problems (That is, people not eating what they were supposed to), but don't cite source to back this up. However, as far as it is known, CALERIE had good adherence (Moreira et al., 2011). Furthermore, what should be obvious: People lost weight, which is an objective, easy to measure metric, indicative of them actually being calorically restricted. Likewise, in the Calorie Restriction Society members, Messrs. Liberman and Reese argue that the sample there was self-selected, disqualifying it. Indeed it was self-selected and it would be a problem if all we did was comparing their health to that of the general population; however the study compares also their pre and post CR biomarkers that are very relevant for cardiovascular disease, showing a remarkable improvement. One could still argue that there is a gene that causes one to both react better to CR and to stay in the CR society, this is what happened in mice after all. But for the CALERIE trial one cannot make this argument; the evidence jointly assessed points to CR being unequivocally effective.
Intermittent fasting
In addition to CR we can also look at intermittent fasting (IF). In the trials above, monkeys could eat within generous spans of time. In IF, feeding windows are limited, and so even for the same caloric intake, the subject will spend longer periods without food, feeling hungry. There is a review (de Cabo & Mattson, 2019) that shows that indeed it is a beneficial regime, above and beyond weight loss. An example is a trial with a group eating 25% less every day, and another group eating normally except for two days a week, where they would eat 500 kcal. Both groups lost the same amount of weight over 6 months, but the IF group had a decrease in insulin resistance, and larger reduction in waist circumference. Some other studies suggest cognitive improvements, but the cardiovascular benefits alone seem reason enough to try.
One option is to skip dinners. If one has lunch at ~1pm and breakfast at ~9am, and ~10 hours are required since the last time of feeding for the fasted state (which corresponds to ketosis) to begin, then one would be in that fasted state for 9-10 hours. In contrast, add a dinner to that at 7pm and you cut the time spent fasting by half.
But on the other hand, there is a recent RCT (Lowe et al., 2020) that shows null results for IF: relative to CR, the evidence for IF is not as strong
Amino acid/Protein restriction
Green & Lamming (2019) is the latest review of the effects of restricting essential amino acids (AR), noting that while we know from experiments and longitudinal studies that there is an effect, the link between amino acid restriction and health-related outcomes remains unclear. The starting point is that perhaps some of the benefits of CR are working through the reduction in protein that goes with CR. Could it be that swapping chicken for chugging olive oil has the same effect as CR? Yes, so it seems.
The amino acids that, if restricted, cause this effect are methionine, and cysteine (The only amino acids containing an atom of sulphur), plus tryptophan . The branched chain amino acids (BCAAs) leucine, isoleucine, and valine seem to have negative effects in promoting insulin resistance (Except in the elderly or athletes, for whom it may be good). However, there is unclear evidence on BCAAs and longevity. BCAA supplementation increases average lifespan in mice, yeast, and worms, but their circulating levels in long lived mice are low. So maybe their reduction good for healthspan, but not lifespan.
Now you may wonder: What foods are high in methionine? In general fish and meat plus eggs. Yoghurt and cheese has less and vegetables and nuts (Except brazil nuts) have the least. For cysteine, that's again meat, but also beans. For tryptophan it's spirulina, fish, cheese, and beans. For BCAA it's again meat, fish, and eggs. So a vegan diet probably works here, as it's cutting out the most substantial sources. Note that these are essential amino acids, so if overdone, one'd die. Methionine in particular is part of every single protein (The start codon encodes methionine).
In humans, there's the case of the Adventists, a religious group whose members don't drink or smoke, and follow various diets (vegan, pescatarian, vegetarian, or omnivore), and so this serves as some preliminary evidence (Orlich & Fraser, 2014), and it does show the effects one would expect from the restriction of animal products (For cancer, diabetes, osteoporosis, hypertension, obesity, and mortality).
This all said, the evidence for AR being effective in humans to promote longevity is weaker than that of CR.
Diet
I have not focused much on the role of diet here. Interpreting nutrition research is hard and it seems that would be its own long FAQ. I am convinced by the arguments presented in the How not to die and How not to diet books that a whole foods plant based diet is optimal. The books discuss what foods to eat, how to cook them, whether supplements work as well as eating their original source and so forth. The usual pitfalls in nutrition research seem to have been averted there: distinguishing between correlational vs RCT studies, trying to find multiple lines of evidence for a result and addressing competing diets.
Rapamycin
Among the longevity-promoting drugs, rapamycin has attracted the most attention: Not only the effect is the largest and robust across gender, but also the same effect has been found in yeast, flies, and worms, in addition to mice. In that regard it is the only drug that has been shown to affect aging across all animal models.
It works by inhibiting mTOR, which in turn upregulates autophagy and possibly suppresses the SASP, increasing the rate of clearance of damaged cellular components. Initially, mTOR inhibits the activity of one of the two components of mTOR, mTORC1, and this drives the anti-aging effect. But chronically administered rapamycin eventually inhibits mTORC2 too, and this causes, among others, immunosuppression (which is what rapamycin is commonly used for), which would make people more vulnerable to infectious diseases.
However, it seems like cycled doses of rapamycin (Or lower dosages) manage to avoid the deleterious effects, while retaining the benefits. There are even some odd results in elderly patients where rapamycin actually boosts the immune system. There is ongoing research on rapamycin analogs, or rapalogs, that may have the benefits of rapamycin without the potential drawbacks.
It also seems (Papadoli et al., 2019) like rapamycin and caloric restriction are substitutes in some cases: Worms where TORC1 is deleted don't enjoy lengthened lifespan when subject to CR. However in other cases (in flies) they may be complimentary.
Rapamycin not only extends life in animal models, it also improves function across various forms of tissue including heart tissue and the central nervous system.
Now, there are those who are even more optimistic about rapamycin. Blagosklonny (2019) for example think the side effects have been overblown and doctors should be more open to it. He cites some work showing greater effects than those found in the NIA trial. Some of those started the treatment earlier than the NIA trial (which gave rapamycin to older mice, however other trials with younger mice also show an effect similar to the NIA trial), and uses a substantially higher dose. The NIA trial itself tried 3 different doses, and the highest one is the one that got the most effect.
But one should be cautious: A meta-analysis by Swindell (2017) finds substantial heterogeneity by sex (females benefited more), and by mice strain. More importantly, the meta-analysis also finds potential publication bias (studies showing more modest effects are not being published). Note that the meta-analysis looked only at mice that had not been deliberately genetically engineered to age faster, as they are trying to see the effect of 'normal aging'. It doesn't include, however, Bitto's, and most experiments have doses way inferior to that paper. The meta-analysis is looking at hazard ratios which may be less intuitive but the collection of papers cited in Arriola Apelo et al. (2016) can give an idea of this dispersion.
Kaeberlein (2014) has a chart of what the dose-response curve might look like (In mice):
One of the trials that Blagosklonny reports (Bitto, 2016) used a dose equivalent to 378 ppm (N=37 in the rapamycin group, ppm is generally mg of rapamycin per kg of weight of the mice), which leads to an increase of 16% in median lifespan, so possibly rapamycin 'saturates' mTOR inhibition and increased dosage beyond a point doesn't help more. Blagosklonny however reports a 60% increase. This figure is in the paper, but corresponds to the increase in life expectancy after the treatment. That is, once mice are middle aged, then they can expect to live 60% longer. But for comparability's sake, the 16% figure is the one we should look at, and this is not too far from the NIA numbers.
Be that as it may and whatever the exact % improvement is, rapamycin is the compound that shows the most promise, working across species, and showing more benefit the earlier the treatment begins. Trialling it in humans would be very interesting. The latest review on mTOR and rapamycin, published just very recently (Liu & Sabatini, 2020) is a good place to find more about this topic.
Acarbose
Rapamycin is very well known in the anti-aging world, but I was surprised to learn that acarbose shows results that match those of rapamycin in males, while showing very weak effect in female mice. Like rapamycin, it's also very cheap. What's the deal with acarbose? The effects were so substantial that the NIA did another, larger trial. In that one, Harrison et al. (2018) report that indeed, the effects were the ones previously found: 16-17% increase in median longevity for males, 8-11% increase in max lifespan (as proxied by 90th percentile survival). They even tried different doses of acarbose, but they all gave similar results. The lower dose was 400 mg per kg.
The way acarbose seems to work is reducing the postprandial (after a meal) blood glucose spike, though it also reduces baseline glucose levels, in particular it stops the release of glucose from complex polysaccharides like starch, and so it also acts as a calorie restriction mimetic.
Acarbose has been used for decades to treat diabetes, and it is only now when the longevity effect has become apparent (in mice! so far). I couldn't really find much more information about it.
Metformin
Metformin is a drug commonly used to treat type 2 diabetes, and it works by activating AMPK, which in turn inhibits mTOR, which in turn should lead to similar effects as rapamycin.
It was not in the table above because it didn't show statistically significant results in the NIA trials. Although has showed promise in other trials, and there is planned trial, TAME, that aims to measure its efficacy in humans. One of the reason some people get excited about Metformin is that it is a safe, cheap, and easily available drug, and it has indeed shown effects comparable to those of rapamycin, but in worms. There are also some studies in mice showing an increase in mean lifespan of 5.83%, though a higher dose was toxic and reduced it by 14.4% (Martin-Montalvo et al. 2013). According to the handbook, in one case it led to an increase in female mice lifespan of 92%, but these mice were not the same mice as in the ITP trials.
Some have argued that metformin has an anti-cancer effect, but this remains debated (Suissa & Azoulay, 2014 for the pessimistic view, Saraei et al. 2019 for the optimistic view). However, as I mention later, it may play a role in combined interventions.
A recent meta-analysis does show health benefits and mortality reduction for humans when comparing diabetics taking metformin with a nondiabetic population (Campbell et tal., 2017)
Piskovatska et al. (2018) say that while the evidence is not strong yet, it tentatively points to metformin potentially having positive effects.
The last review on metformin is Glossman & Lutz (2019) is more mixed, and looks at RCTs and observational studies (As in the Campbell meta-analysis), noting that metformin's effects.
The rationale for the ongoing or planned metformin trials is almost exclusively based on observations (associations) of potential benefits in a diabetic (or prediabetic) population. Its efficacy even in an at-risk cohort of aged people has not yet been proven.
All things considered, there is support for continued trials of metformin in humans, and there is some evidence that it could improve diseases associated with aging; however we still don't know if it would help with increasing maximum lifespan in humans.
Senolytics
This is a list of drugs that have potential to clear senescent cells (From Shetty et al. 2018). Unity Biotechnologies is also developing a few more and so is Oisin Biotechnologies. The idea is simple: If, as explained earlier, senescent cells accumulate and this is negative due to the SASP, let's just get rid of them.

Some early work here is the Baker et al. (2011) paper showing reduction in sarcopenia, adipose tissue, and cataracts in the treated mice (They didn't look at lifespan). Later, Baker et al. (2016) shows that in addition to improving health, it also extends median lifespan by 25%, but it failed to increase max lifespan in one of the two strains of mice they looked at. In the other, the effects are, if anything, small. Xu et al., 2018, seen earlier, showed increase in lifespan after treating mice with quercetin and dasatinib (A senolytic drug combination). Intriguingly, treated mice show a higher mortality rate at the end of their life. This is a similar pattern as seen elsewhere: If median lifespan increases but maximum does not, it means aging must be accelerated near the very end. One can of course find studies that do also show a small increase in maximum lifespan, not with quercetin but with fisetin (Yousefzadeh et al., 2018).
I could find at least one person arguing that there may be longer term downsides, the argument being that if senescent cells are killed, neighbouring cells will replicate to replace them, shortening their telomeres, in turn inducing more senescent cells. On this view, senolytics are ineffective at best and negative at worst ( Fossel 2019). This is a very minority view - the majority being that there are no major downsides to senolytics and here's a critique of the argument just presented.
Another avenue to treat senescent cells is to let them be, but modulate the SASP so that it stops disrupting nearby cells, using so-called senomodulators, or senostatics (Kang, 2019), one of them being rapamycin. Notably, the blind mole rat Spalax, which like the more well known naked mole rat is very long lived, also have senescent cells, but their cells do not have an inflammatory SASP! (Odeh et al., 2019).
Antioxidants
Antioxidants have not been shown to extend max lifespan, although they may extend mean lifespan, at least in wild-type animals. A Cochrane review of the evidence from RCTs in humans (Bjelakovic et al., 2012) showed no effect, or even negative effects from (excessive) antioxidant intake. Vaiserman et al. (2016) show that while most studies in flies or worms do show positive effects, in mice the evidence is weaker. The review suggests that perhaps additional antioxidant intake downregulates endogenous antioxidant production, for a net effect of zero. This makes sense under the model that ROS may also act as signaling: You want some ROS but not zero ROS, so in experiments where there is excessive antioxidant supplementation have actually worsened health and reduced lifespan in animal models.
Applying that idea [antioxidant supplementation] to humans, however, appeared rather problematic. Most clinical trials failed to show any clinical benefits of antioxidant therapy for aging-related disorders such as cardiovascular disease (Kris-Etherton et al., 2004; Sahebkar et al., 2015), stroke (Schürks et al., 2010), and type 2 diabetes (Sheikh-Ali et al., 2011; Suksomboon et al., 2015). This controversy could likely be related to the excessive antioxidant intake or to insufficient antioxidant bioavailability in relevant organs to reduce oxidative damage (Doss, 2012). The excessive levels of exogenous antioxidants can disturb endogenous signaling mechanisms and thereby might be deleterious (Halliwell, 2011). So, the use of multivitamins more than seven times per week has been demonstrated to be related to a significantly enhanced risk for prostate cancer in comparison to never users (Lawson et al., 2007). Dietary intake of vitamins C and E combined precluded the health-promoting effects of exercise in the Ristow et al. (2009) study. [...] Some authors claim that the oxidative damage theory of aging is fundamentally wrong, though this has been a topic of intense debate within the field over the past decade. This theory, however, still has many adherents and needs to be decisively verified in further studies.
Sirtuin Activating Compounds
Sirtuin activating compounds (STACs) increase the activity of sirtuins. They are usually paired with NAD boosters (Most commonly, nicotinamide riboside (NR) less commonly nicotinamide mononucleotide (NMN)), to get extra NAD+ to fuel the increase in sirtuin activation.
Resveratrol (Which also happens to be an antioxidant, an AMPK activator, a PARP1 activator, and more) is the most popular one, acting as a SIRT1 activator. As mentioned above, the ITP found no effect of resveratrol in their mice, although it seems to work in worms, yeast, and flies. A recent review from Bonkowski and Sinclair (2016) point to (and note that the Sinclair lab focuses on sirtuins) some studies that found positive outcomes, although early work was flawed (A fluorescent moeity, moeity being just a small molecular add-on to a larger molecule, was thought to cause the seeming pattern of activation, rather than the STAC itself), later work showing that the fluorescent add-ons were actually not necessary and that resveratrol indeed activates sirtuins.
Resveratrol is also present in red wine, however the concentrations are so low that you would need to consume thousands of litres to reach a therapeutic dose (Weiskirchen & Weiskirchen, 2016)
Resveratrol administered to primates for two years improved their cardiovascular health, but the monkeys were being fed a high sugar, high fat diet, so if this holds in humans, it may not translate to a healthier population. Various studies are cited in the review that were done in humans with some success for obese men, but none for non-obese men; Table 2 of the Bonkowski-Sinclair paper shows some trials of resveratrol for the treatment of particular conditions, but not lifespan. Overall it seems to me the review is too optimistic and so I searched for additional evidence. Back in 2011, the Resveratrol2010 conference formed a working group to assess the evidence (Vang et al., 2011), rating it as inconclusive, but promising. Fast-forward to more recent times and Berman et al. (2017) mentions positive effects for certain kinds of cancers, Alzheimer's or diabetes. Fogacci et al. (2018) 's review finds that resveratrol improves cardiovascular health in diabetics or obese individuals.
Does it really work, despite the ITP finding a null effect? Was it that resveratrol makes you healthier but not live longer? Maybe the ITP study had a low dose. Miller (2010) fed the mice 200 mg/kg of resveratrol. In humans, that dose would be 16 grams, which seems high, as David Sinclair himself takes 0.5 gram of resveratrol a day. So clearly the issue wasn't that the dose was low. Perhaps that it has to be accompanied with NR or NMN? There are no studies on this. Resveratrol is more easily absorbed when taken with fat (Sinclair takes it with yoghurt), but 32x the dose Sinclair takes, in mice, seems like it should be sufficient.
Other STACs like SRT1720 or SRT2104 do work on regular mice fed a regular diet, but they haven't been as intensively tested as resveratrol. As for NAD precursors (NR or NMN) supplementation, this would help all sirtuins. These also seem to work for improving health outcomes, but again the evidence on lifespan is less certain (Qian & Liu, 2019).
David Sinclair, in an interview with Peter Attia discusses the failure of the ITP study and doesn't seem surprised, arguing that you really do need the fat.
Others of these STACs are pterolstilbene, butein, or quercetin.
[Edit 2022-03-19] Since I first wrote the FAQ I've grown more skeptical about NMN and resveratrol. See here.
Parabiosis
This (Eggel and Wyss-Coray, 2014) is probably one of the most well known, due to the peculiarities of the process involved: It consists of transfusing blood or plasma from younger individuals to older ones, this in particular is known as heterochronic parabiosis (Ludwig & Elashoff, 1972), to distinguish it from cases where the animals are the same age. In early experiments, this was done by fusing the circulatory systems of two mice of different ages.
Later, in 2005 and then 2011, other experiments with a similar design showed that the older mice of the pair showed improved tissue regeneration, heart function and cognitive performance, while the reverse was observed in the younger mice. The reason this was happening is that there are proteins or other elements in the bloodstream of old mice that are harmful in higher concentrations like CCL11 while others that are beneficial are reduced in amount (Conese et al., 2017)
Of course, we all wonder if this work in humans. The TV series Silicon Valley made popular the concept of blood boys, which ended up being memed into reality by real-life startup Ambrosia, who provided plasma transfusions from young donors. They claimed it had benefits for their patients after a trial, but they never released any data, and the after the FDA issued a warning saying that the treatment is not effective, they temporarily closed down, but they have since resumed business.
There is no evidence indeed that this works in humans, but there are ongoing trials to see if it does work. The one I could find is the PLASMA (Plasma for Alzheimer Symptom Amelioration) study at Stanford. So far they have merely done an RCT in a very small sample (9 people per group) to prove that the procedure is safe (Sha et al., 2019). This all sounds somewhat disappointing, especially given that the study was completed in 2017 and back then it was reported that in that same data there was no improvement in alzheimer. This work seems to have done in part by Alkahest, a company founded by Wyss-Coray.
More recently, Horvath et al. (2020) showed that more extensive replacement of blood plasma (Rather than joining mice, infusing them with a proprietary plasma formula every few days) leads to improvements across the board with the exception of the brain. The authors speculate that this may be because
This, of course, raises a related and equally interesting question, which is why does plasma fraction treatment not reduce brain epigenetic age by the same magnitude as it does the other organs? We can only begin to address this question after having first understood what epigenetic aging entails. As it stands, our knowledge in this area remains limited, but it is nevertheless clear that: (a) epigenetic aging is distinct from the process of cellular senescence and telomere attrition 41, (b) several types of tissue stem cells are epigenetically younger than non-stem cells of the same tissue 42,43, (c) a considerable number of age-related methylation sites, including some clock CpGs, are proximal to genes whose proteins are involved in the process of development 44, (d) epigenetic clocks are associated with developmental timing 22,45, and (e) relate to an epigenomic maintenance system 20,46. Collectively, these features indicate that epigenetic aging is intimately associated with the process of development and homeostatic maintenance of the body post-maturity. While most organs of the body turn-over during the lifetime of the host, albeit at different rates, the brain appears at best to do this at a very much slower rate
We don't know what their proprietary formula is, but perhaps it's just (or mostly) fresh albumin. Most circulating proteins are albumin, so getting that fixed is a large leverage point. There have been preliminary trials in humans showing cognitive benefits of infusion with fresh albumin (Loeffler, 2020) and work relating the oxidization of albumin to aging (Luna et al., 2016) exists.
On the other hand, the Conboys have also two recent papers (Mehdipour, 2020; Mendipour, 2020) where they merely dilute the plasma, replacing plasma with fresh albumin. The interpretation of the paper is that diluting "junk" that's present in blood is beneficial and they try to justify that it is not the additional albumin they are adding, but they don't go as far as claiming that the albumin has no effect ("Ectopically added albumin does not seem to be the sole determinant of such rejuvenation").
Both the SENS foundation and Eli Dourado take these results to imply that the benefits from parabiosis / plasma exchange are not due to anything beneficial being present in young blood, but rather to the accumulation of "junk" in old blood. SENS does point to a bunch of specific cases where putative "young blood factors" turned out to be not so, ultimately GDF11 when tested on on lifespan has no effects. But they don't point to a study where they tried to inject young blood to old mice or other models; after all it is perfectly possible that GDF11 is not effective yet something else is. The only such study I could find was Shytikov et al. (2014) where they injected young blood plasma to old mice, finding no lifespan effects. The bulk of the evidence then points to dilution being a robust effect whereas we have not identified any specific factors in young blood that can induce rejuvenation. What then, is in Horvath's mysterious proprietary Elixir? Whatever is it, it doesn't have to be -nor they claim it to be- based on or inspired by, young blood factors. They have just shown that there are injections one can administer that cause systemic rejuvenation effects.
A clean experiment to determine the relative contributions of each [using the same assays] has, to this date, not been done yet.
Epigenetic cellular reprogramming
So the epigenome gets damaged over time. But we also know that somehow it gets more or less reset (Not perfectly, this is why transgenerational epigenetic inheritance is a thing) when the zygote forms (Rando & Chang, 2012 so there should be a way to take a cell and reset it. Back in 2006, Shinya Yamanaka showed that one could take a random cell, make it express four genes (The initials of which are OSKM, or elsewhere known as Yamanaka factors) and make that aged old cell into a fresh and young stem cell.
Now, if you just did that to an organism it would become a blob of stem cells, and indeed something like this is what happens if one happily forces expression of OSKM in mice, one gets cancer and teratomas. But what if instead one goes only halfway? Using genetically engineered mice that express OSKM only when dosed with the antibiotic doxycycline, and having the mice on and off that drug, Ocampo et al. (2016a) rejuvenated and extended the lifespan (maximum by 20% and median by 33%) of genetically engineered mice that show premature aging. Seeking a less carcinogenic combination of factors, the Sinclair lab (Yuancheng et al., 2019) tried just OSK in the eye of mice to see (no pun intended, I promise) if they could regenerate the optic nerve after an injury, and restore vision in mice. And they did it.
There are many ways to reprogram cells: Some are "integrating" (They cause permanent edits in the genome) like various kinds of virus, while others are temporary, like plasmids, adenoviruses (Unlike other virus, these just inject their DNA into the cell to replicate, rather than changing the host's DNA), proteins or small molecules. The best one in terms of efficiency and not being permanent is the Sendai virus; which however is expensive and hard to produce (Ocampo et al., 2016b).
Oddly, senescent cell's SASP make it easier for neighbouring cells to be reprogrammed, so perhaps reprogramming followed by senolysis would have synergistic effects (Ofenbauer & Tursun, 2019).
Out of all the therapies I've reviewed, with no doubt this one seems the most promising to me. There is a startup, Turn.bio that is trying to address all hallmarks of aging through reprogramming.
[Edit 2022/03/31]: Since writing this there are a few more companies pursuing this: Calico, NewLimit, or Retro.
Read a longer review of partial reprograming here.
Combined interventions
After reading the above, you probably thought: Hey, shouldn't we just try everything at the same time and see what happens? Indeed this seems to work.
We said that metformin doesn't seem to do much and that rapamycin does. But what if you combine the both? Well, rapamycin leads to 10-13% longer median lifespans in mice at 14 ppm. Combined with metformin (1000 ppm), that goes up to 23%, even when metformin on its own had no effect (Strong et al., 2016). It reminds to be seen, however, what happens with the high dosages of rapamycin than on its own are already able to achieve similar effects.
In flies, a combination of rapamycin, trametinib, and lithium extended their life by 48%, which is more than the sum of the effects of each of them(Castillon-Quan et al., 2019).
In worms we already knew that certain mutations along the mTOR or IIS pathways can make worms live substantially longer. But what if we create a super-worm with many of them? Yes, they live longer (500% more) than what one would expect from the sum of the effects alone, report Lan et al. (2019), though again this same combination is unlikely to have as large effects in longer-lived species.
This doesn´t mean that every combination is good. A recent study (Palliyaguru et al., 2020) showed that combining a STAC with metformin reduces longevity even when, in that study, metformin on its own has no effect and the STAC increases median (but not maximum) lifespan. More work is needed to determine exactly when drugs conferring putative longevity benefits will lead to synergies or the opposite.
Conclusion
Longevity research is an exciting area that has been making great progress in recent years. From the early discoveries that ageing can be modulated to the current advances in understanding how aging works, and how therapies could be developed to live longer, healthier lives.
Progress seems easier on the "healthier" side of things, with many of studies showing that it is easier to prolong healthspan or expected lifespan and cure certain conditions that occur in the old age rather than the maximum lifespan of our species. Current research seems like it could enable most people to live past 100 years in reasonably healthy conditions, a feat that, to a lesser degree, is accomplished today by a tiny fraction of supercentenarians.
What about drastically extending lifespan? While indeed it has been shown that this is possible in animal models, it remains to be seen what could work for humans. Recent work in long lived mammals such as the naked mole rat at Calico will help us understand how animals more similar to us could achieve negligible senescence.
Something that seems a constant along this research is that most of the focus is on investigating the diverse hallmarks and therapies to address them, thus one finds that there are telomere researchers, mTOR researchers, sirtuin researchers and so on. However, if all of this is tightly linked, fixing one of the factors won't on its own be shown to have a substantial effect, and this is what the limited research on combined interventions show
Questions for further analysis
- SENS: Their history, achievements, and approach. SENS started as a controversial research project and their explicit aim is to extend maximum lifespan.
- Adaptations in long lived species: How they do it to live that long.
- Plants very rarely get cancer (Doonan & Sablowski, 2010), and many of them live very long. How does this work? Can anything learned there be applied to animal cells? Probably not: Our cells are not covered in cellulose (Which would stop cancer fro, spreading), and plants are more redundant than us, being built of simpler, more repeated parts.
- The entrepreneurial and investing side: What are startups working on, what are VCs funding?
- Aging processes across species. Are the key drivers, or kinds of damage different in humans vs, say, worms.
- What are the limits of reprogramming? Something not discussed here but that is another kind of damage concomitant to aging is the accumulation of lipofuscin, undigested leftovers from the digestion of cellular garbage. Another is glucosepane crosslinks in the extracellular matrix. Do rejuvenated cells also help clear out this?
- What's the relative importance of reducing damage (What most of the research is focused on) and letting it happen, but fixing it (What SENS is focusing on)
Changelog
- 2020-01-20
- Fixed typos
- Clarified definition of epigenetics
- Included effect of retrotransposons in aging
- Clarified role of sirtuins (I had said PARP depletes NAD, but sirtuin activation too also depletes NAD, along with overexpression of CD38)
- A study I cited that said sirtuin overexpression in flies does not increase lifespan turns out to be not correct as it overexpressed them too much. Later work with more modest expression does find lifespan gains.
- Added an explanation for the contextual role of SIRT1 as a repressor or promoter of tumors
- Added the example of the Adventists as some evidence of the effects of a a low-meat diet
- Corrected the sample size for the Bitto et al. study, I said it was N=17, it was N=37
- Removed section on glycemic load, added a study that shows some skepticism
- Added example of study that used only the OSK factors to regrow the optic nerve and restore vision in old mice
- Added warn that gene knock-out can have unexpected effect and thus confound studies
- Added link to SSC post on calorie restriction
- I had said that metformin activates AMPK through the insulin pathway. Rather, it activates AMPK directly (What was I thinking)
- Added some quotes
- More optimistic conclusion for the STAC section
- More optimistic take on metformin
- 2020-01-21
- Added paper on telomere shortening rate
- Added definition of RNAi
- Note that the finding that there are stem cells in the brain is recent
- Caveated the applicability of the Gompertz model in humans
- Rewrote Hallmarks's epigenetic section
- Warned that the lack of effects in mice doesn't imply that it won't work for humans.
- Added link to Gwern's cost benefit analysis of longevity interventions
- Defended Caloric Restriction Society study
- 2020-01-22
- Role of DNA damage was underplayed in original, remarked that long lived species have enhanced DNA repair.
- Added comment on the role of the immune system in cancer
- Stressed the importance of combined therapies in the conclusion
- Mentioned possible role of telomere shortening on sirtuin inhibition
- 2020-01-23
- Mentioned acetylation in the epigenetics section
- Noted that the hypomethylation pattern that goes with aging might not be true due to some fresh unpublished work
- Noted that some cells don't share the same DNA as others (In a deliberate way, as part of the immune system)
- I said that chromosomes are not in the nuclei; rather they are not visible because they are uncoiled.
- Various issues around the explanation of the daf-2/IGF-1 paragraph in the History of Aging research section
- Typos, more typos.
- 2020-01-24
- Two extra papers on the evidence behind cancer and the immune system
- 2020-02-02
- The generalised hypomethylation pattern does not seem to be there, added a section in Measurement Issues
- 2020-02-04
- Added sections on mitochondria and inflammation
- SENS to Hallmarks table
- 2020-02-06
- Typos
- Flipped the telomere sequence to TTAGGG
- Better explanation for the Hayflick limit and the role of telomeres.
- 2020-02-19
- Extra paper trying to explain why long telomere mice live longer
- 2020-03-07
- Typo fixing
- 2020-03-26
- Various fixes from here
- 2020-06-08
- Turns out Ambrosia is back in business! Also they were not forced to shut down by the FDA, they voluntarily closed down for a bit
- 2020-06-09
- Mentioned that the monkeys in the NIA study were genetically diverse which might introduce a downward bias in the effect observed
- Added link to epigenetic clocks post
- 2020-06-23
- Added zebrafish with short telomeres paper
- Fixed typos
- 2020-06-29
- Added new bad synergy paper
- 2020-07-10
- Mentioned that CD38 is the main culprit for NAD depletion
- Added Sinclair interview discussing ITP
- 2020-07-25
- Added link to immunosenescence review
- 2020-10-06
- Added link to new IF study
- 2021-02-21
- Added Conboy plasma dilution experiments, commented on the relative importance of plasma dilution vs young plasma factors
- 2021-03-01
- Added link to What is Aging essay
- 2021-03-10
- Added Vijg paper about DNA mutations in aging
- 2021-07-26
- Typos
- 2021-08-08
- Reorganized titles
- 2021-08-30
- Added "diet" section
- 2021-09-19
- Added remarks on genetically diverse mice. A paper claiming diverse strains suffer negative effects is suspect.
- 2021-10-30
- Added paper that did the Liao one with a larger sample, as expected the negative effects go down
- 2021-11-08
- Added paper with evidence against DNA damage, in particular mutations, being a major driver of aging
- 2022-03-31
- Linked to new summary of partial reprogramming
- 2023-09-04
- Added Derek Lowe post on sirtuins
Citation
In academic work, please cite this essay as:
Ricón, José Luis, “The Longevity FAQ”, Nintil (2020-01-19), available at https://nintil.com/longevity/.
Backlinks
- Aging is already solved in vitro. What comes next?
- Crypto promises, crypto hype
- Notes on 2020
- Epigenetic clocks: A review
- How to solve aging
- Immunosenescence: a review
- Images and Words: AI in 2026
- Kenneth Arrow on the welfare economics of medical care, a critical assessment
- Beyond lifespan as a metric in aging research: why reprogramming is promising
- Links (67)
- Longevity — what we know so far (video)
- Is cellular senescence irreversible?
- The Great Stagnation: A misleading narrative
- [Guest post] How Substack Became Milquetoast
- Telomeres: everything you always wanted to know
- US Federal R&D spending decline: a breakdown
- What should you remember? SRSing your life
Comments