Immunosenescence: a review
Introduction
This post reviews the changes that occur in the immune system with age, and why that might be. Usually I aim to provide more or less coherent, first-principles explanations that capture what is the state of the art in the relevant field, figuring out if something is a collection of heterogeneous findings, just noise, or a true simple fact hidden under imperfect study designs. I have not been able to do that here, as I found a lot of discrepancies in the underlying literature, some of which have also been presented here as illustrative. It is unsettling when you see recent literature reviews claiming the opposite things as a settled fact. Normally it’s possible to find later work that explains why there is a disagreement, or specific papers that try to deal with them through superior experiments. In any case, this review is still useful to get a general idea of what happens to the immune system with aging. Rather than a coherent story with clear villains and heroes, this is more of a summary of around a month of exploring this topic. I think given more time and narrower questions it should be possible to have more definitive answers.
To some extent the origin of this review was to examine the role of the thymus in immunosenescence, and in particular to assess claims that a lot of cancer could be prevented via thymic regeneration, and so a lot of space has been devoted to that. Being a post about the immune system, it may be surprising to see little here about infectious disease, and while there is a link to a specific review on that for the elderly, I also wanted to focus on the “best case” scenario by defining infectious disease away from the scope of the review. This is because another goal of this review is more to identify the set of causes of immunosenescence, rather than focusing on understanding the complex network of interactions that occur in the immune system. The two culprits I expected at the beginning of the review to be so were indeed so, perhaps to a lesser extent than I initially assumed: the thymus and hematopoietic stem cells (HSCs), although the latter not so because they get damaged but because their microenvironment changes. As for why this is, I thought it would make the review even longer and so I left it there. The factors I did not have immediately in mind, aging of the spleen and lymph nodes are there but were infrequently discussed in the literature. One of the things I thought would have more of an impact was infection with cytomegalovirus. However, after reading the literature I ended up with the impression that the evidence is not that clear, and if anything, it is a secondary characteristic associated with age but not a core concept within immunosenescence.
Inflammation is a term that appears a lot, not just here but in the gerontology literature in general. it seems it’s used as a sort of “magic word” that can always be blamed for something bad, or where you can connect the effect of whatever is it that you are studying. To hyperbolise a bit, everything causes inflammation and inflammation causes everything. This is not very helpful. Probably the only way to really understand inflammation in the context of aging is doing a review solely focused on that, seeking to answer questions like what are sufficient or necessary conditions to restore inflammation to youthful levels? What are the feedback loops and dynamics involved? That would help both design experiments and design better therapies.
It won’t sound surprising to say that the immune system degrades with age. Looking at the rates of death from infectious disease clearly reveals an association with age, as an example here’s the flu and Covid-19, clearly showing a marked increase with age: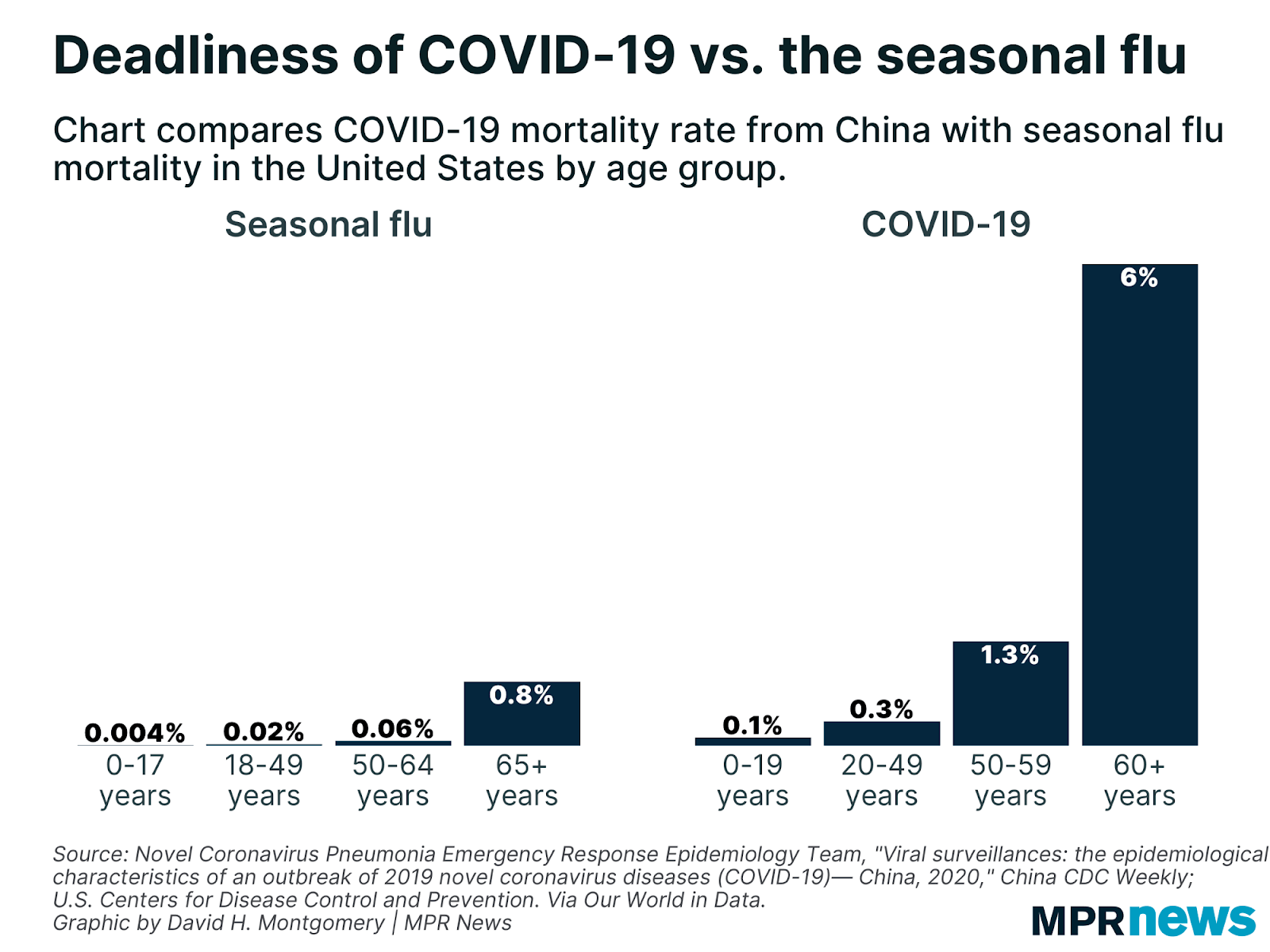 The same is also true for atherosclerosis, diabetes or autoimmune diseases, in all of which the immune system plays a role. Of course it’s hard to disentangle the immune system from the rest of the body, but looking at specific measures of immune system health such as the ratio between lymphocytes and monocytes (LMR) yields powerful predictors of survival for heart failure (Silva et al., 2015), various kinds of cancer (Szkandera et al., 2013, Hu et al., 2014, Eo et al., 2016, Ma et. al 2018), arthritis (Du et al., 2017), and of course infectious disease (Piotrowski et al., 2020, Wang et al., 2019, Liu et al., 2020). This ratio goes down with age, chiefly by a reduction in the number of circulating lymphocytes which occurs very early in life.
The same is also true for atherosclerosis, diabetes or autoimmune diseases, in all of which the immune system plays a role. Of course it’s hard to disentangle the immune system from the rest of the body, but looking at specific measures of immune system health such as the ratio between lymphocytes and monocytes (LMR) yields powerful predictors of survival for heart failure (Silva et al., 2015), various kinds of cancer (Szkandera et al., 2013, Hu et al., 2014, Eo et al., 2016, Ma et. al 2018), arthritis (Du et al., 2017), and of course infectious disease (Piotrowski et al., 2020, Wang et al., 2019, Liu et al., 2020). This ratio goes down with age, chiefly by a reduction in the number of circulating lymphocytes which occurs very early in life.
This should bring to mind the thought that these are just correlations, how sure can we be that these are causal. Interventions that aim to boost the immune system do not only cause a change in the raw numbers in lymphocytes or monocytes. Other than cloning the host’s white blood cells and injecting them back to force a desired ratio, and doing so periodically, interventions will most likely be affecting something else; and likewise unrelated interventions may affect the LMR via some other mechanism.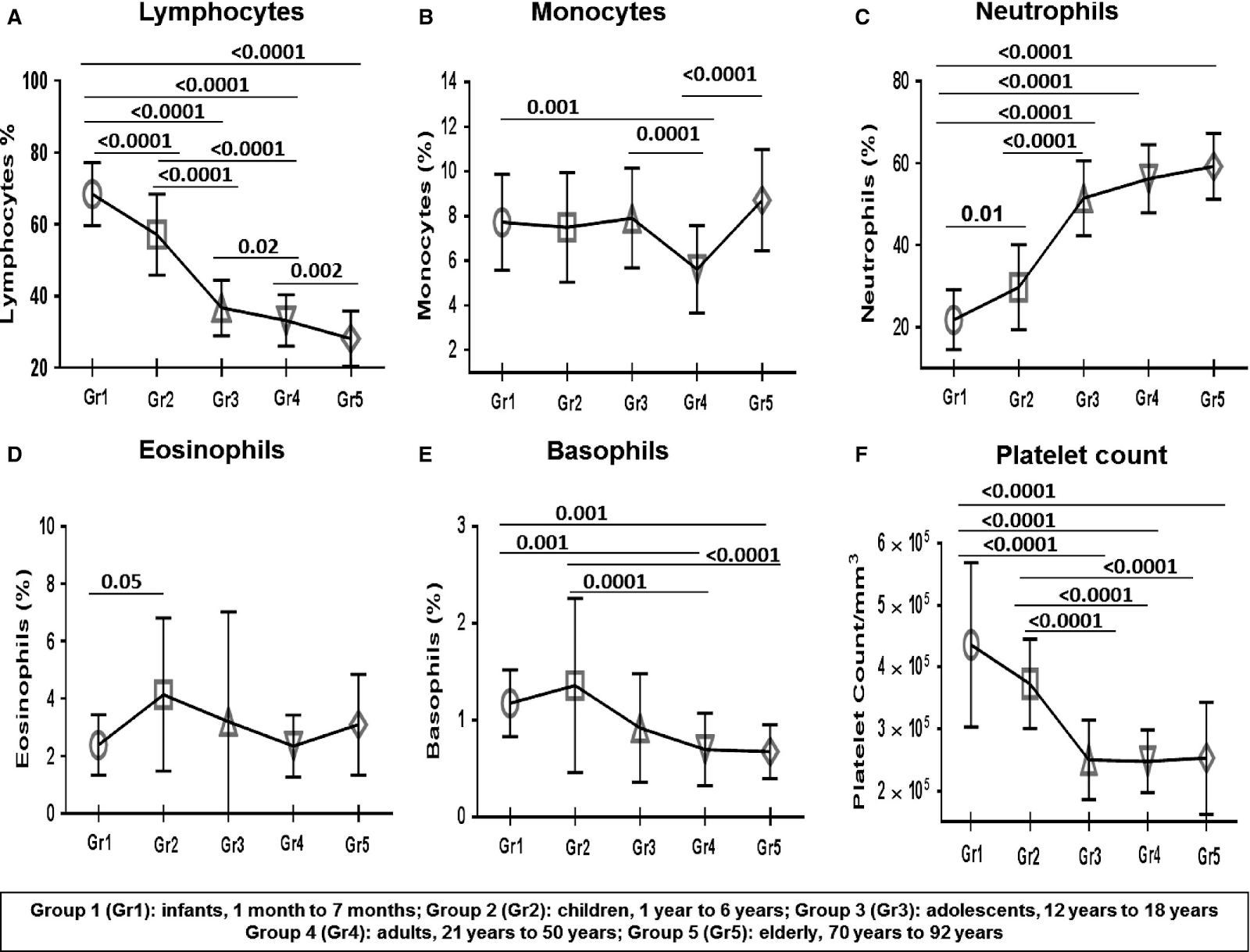
The immune system
It’s worth going over a few of the different subpopulations that comprise the immune system. This list is not exhaustive, and in fact new kinds of cells are discovered every year.
- Lymphocytes / Lymphoid lineage
- Adaptive immune system
- T cells
- CD4 Helper T cells. Contribute to CD8 T cell activation
- CD8 Cytotoxic T cells. These are the ones that, once co-stimulated by B cells will kill target cells by breaching their cell walls with perforin, then injecting granzymes, which trigger apoptosis.
- Regulatory T cells (Tregs). These have an anti-inflammation role, and will disable CD8 cells
- Natural Killer T cells
- CD45RO Effector/Memory T cells (Temra)
- Virtual memory T cells
- Gamma-Delta T cells
- B cells. They are able to recognize antigens and produce antibodies.
- T cells
- Innate immune system
- Natural Killer (NK) cells. Kill (some) infected and cancerous cells.
- Adaptive immune system
- Myeloid lineage / Innate immunity system
- Professional phagocytes
- Monocytes. They differentiate into macrophages when they migrate into tissues. Alternatively they can become dendritic cells, presenting antigens for the adaptive immune system to pick up
- M1 macrophages. Generate inflammatory cytokines and help attract other immune cells
- M2 macrophages. They serve an anti-inflammatory role
- Neutrophils. They phagocyte, and capture pathogens with NETs , these are the most common type of white blood cell
- Monocytes. They differentiate into macrophages when they migrate into tissues. Alternatively they can become dendritic cells, presenting antigens for the adaptive immune system to pick up
- Non-professional phagocytes (They can phagocyte, but that’s not their main role)
- Basophiles
- Eosinophils
- Professional phagocytes
Again this is not a full picture, for example NK cells can also phagocyte besides attacking cancerous or infected cells.
A longer list of markers associated with each T cell can be found in Van den Broek et al. (2018) which includes the somewhat ominous:
Although different laboratories have their own ‘favourite’ naive T cell markers, most of these actually select for highly overlapping pools of naive T cells… However, it is important to realize that this naive T cell identification system will include small populations of functionally distinct T cell subsets, such as forkhead box protein P3 (FOXP3)-expressing regulatory CD4+ T (Treg) cells, stem cell memory T (Tscm) cells and memory T cells with a naive phenotype (BOX 1). Conversely, memory-phenotype T cells that may be antigen-inexperienced, such as virtual memory T cells and innate memory T cells, are excluded”
The same is true for Temra cells, that reexpress one of the markers present in naive cells, CD45RA. This is important to keep in mind when looking at works that aims to find this or that population. Maybe they will be also counting part of other population too, biasing the analysis. All of these cells ultimately come from hematopoietic stem cells (HECs) in the bone marrow, from where they are released to the blood and differentiate into all the different types. Lymphocytes are special in that they can increase their numbers by clonal expansion to more effectively react to a given pathogen.
T cells in particular are so named because after they are generated in the bone marrow as thymocytes, they migrate to the thymus to be trained before they differentiate into a specific type of cell. In the thymus they are selected so that they don’t attack healthy host cells. Thymic epithelial cells (TECs) express all sorts of genes that produce various markers in the TEC’s surface which a T cell has to recognize or else it will be forced to undergo apoptosis. In the thymus they undergo V(D)J recombination, generating a large diversity of T cell receptors (TCR), with each individual cell having 1 type. To be able to mount an effective response to a pathogen, there have to be T cells that recognize molecules that are on the surface of the pathogen (antigens), these cells can either be “effector” or activated T cells that have seen the pathogen before, or “naive” T cells (expressing CD45RA) that require activation from antigen-presenting cells like dendritic cells to start cloning themselves and mount and immune response.
Once their training is finished they are released and circulate mainly around the lymphoid organs such as tonsils, spleen, or lymphoid nodes. Only 2% will be present in the blood (Brahma et al., 2018, Farber et al., 2014), and the proportion of different kinds of T cells will be different in various regions of the body, for example there are more naive T cells in blood than in the spleen (Thome et al., 2014) thus looking just at the blood will give an imprecise view of total T cell diversity. With age, a striking change that occurs is the loss of naive CD4 and CD8 T cells, a change that is more marked for CD4s outside of the blood. Conversely, CD8 Temra cells accumulate with exposure to cytomegalovirus (and most people are so exposed). This latter point will be discussed later.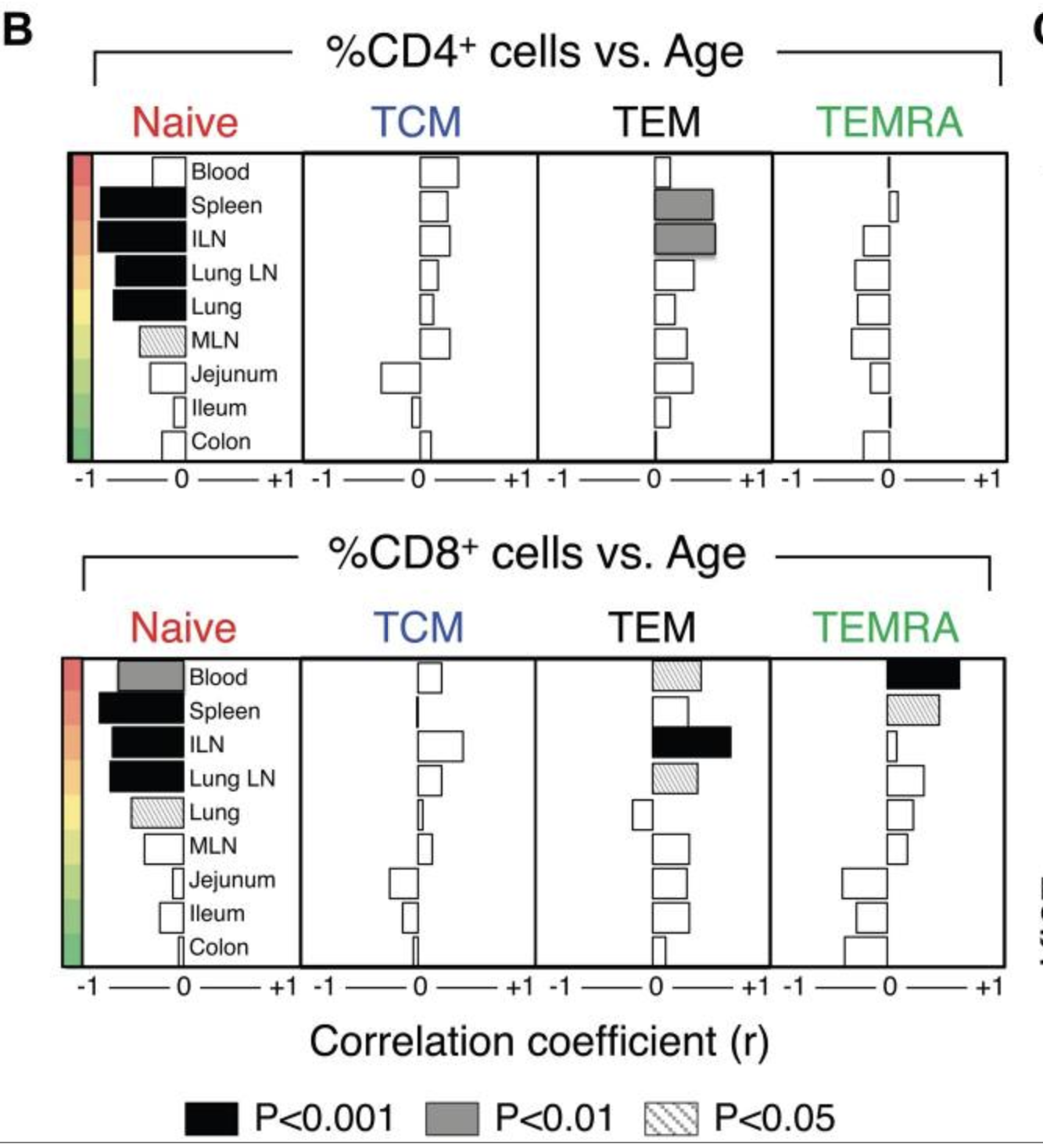
Interleukins and assorted molecules of interest
The immune system cells regulate their activity by, among others, the exchange of cytokines. They don’t have a single role, but it is useful to explicitly name some ones and their common purposes.
- IL1b, IL6, TNFa: markers of inflammation
- IL7: Promoter of TEC development. Secreted by the bone marrow (to commit cells to the lymphoid lineage) and thymic epithelial cells to induce maturation of thymocytes
- IL-21: Typically produced by T cells to induce proliferation in fellow T cells, and turn macrophages into an inflammatory M1 phenotype
Immunosenescence
Immunosenescence is the progressive degradation of the immune system. As an overall theme, it seems like part of the immunosenescent phenotype is not the result or damage or exhaustion, but of a program designed to save on energy and better adapt to recurrent threats, as opposed to new ones (Shanley et al., 2009). Indeed, looking at elderly individuals, one finds reduced counts of adaptive immune system cells, a shrinkage of the energetically expensive thymus, and the innate immune system expanding to cover for the retreat of the adaptive immune system. Chronic, systemic, inflammation is also commonly seen in the elderly, and this may represent a continuously alert innate immune system, some of it potentially being an adaptive response (Fulop et al., 2018). This all might make sense if the organism doesn’t encounter new pathogens, as there will be plenty of memory T/B cells that will recognize the antigen. However for new pathogens the situation is more dire. Evolutionarily, the argument has been made that this is exactly the situation encountered by our ancestors, who wouldn’t have travelled great distances or lived in a highly populated world that allows fast transmission of pathogens, and they wouldn’t have lived long enough for cancer to be a big issue anyway.
Other lymphoid organs also undergo this involution, like the spleen or the lymph nodes, but perhaps for different reasons. This is an area I didn't spend much time on during the review.
Another component of immunosenescence is not regulated, but accumulation of damage, in particular epigenetic noise in the hematopoietic stem cells, leading to less effective immune cells in general, not just lymphocytes but also macrophages. Circulating T cells will also eventually undergo cellular senescence after rounds of clonal expansion, preventing them from further replicating, and generating the inflammatory SASP.
Not everyone agrees that HSCs and the thymus are key causes of aging: Goronzy & Weyand (2019) rather point to the existence of plenty of immunocompetent elderly as a counterexample; for them the key goal would be to reduce inflammation, and that should be sufficient. This is an odd response, because an example does not disprove a trend.
While this review largely has vertebrates in mind (invertebrates don’t have an adaptive immune system, and more primitive innate immune systems, so they are less useful to understand the case of humans), immunosenescence also occurs in these cases, an example being C. elegans (Youngman et al., 2011, McHugh. 2019) where the process is driven by mechanical damage to the intestine (which facilitates colonisation by bacteria), loss of the intestine itself due to runaway autophagy (Ezcurra et al., 2018), and general loss of proteostasis as it is programmatically downregulated to free up energy for reproduction (Kikis, 2016).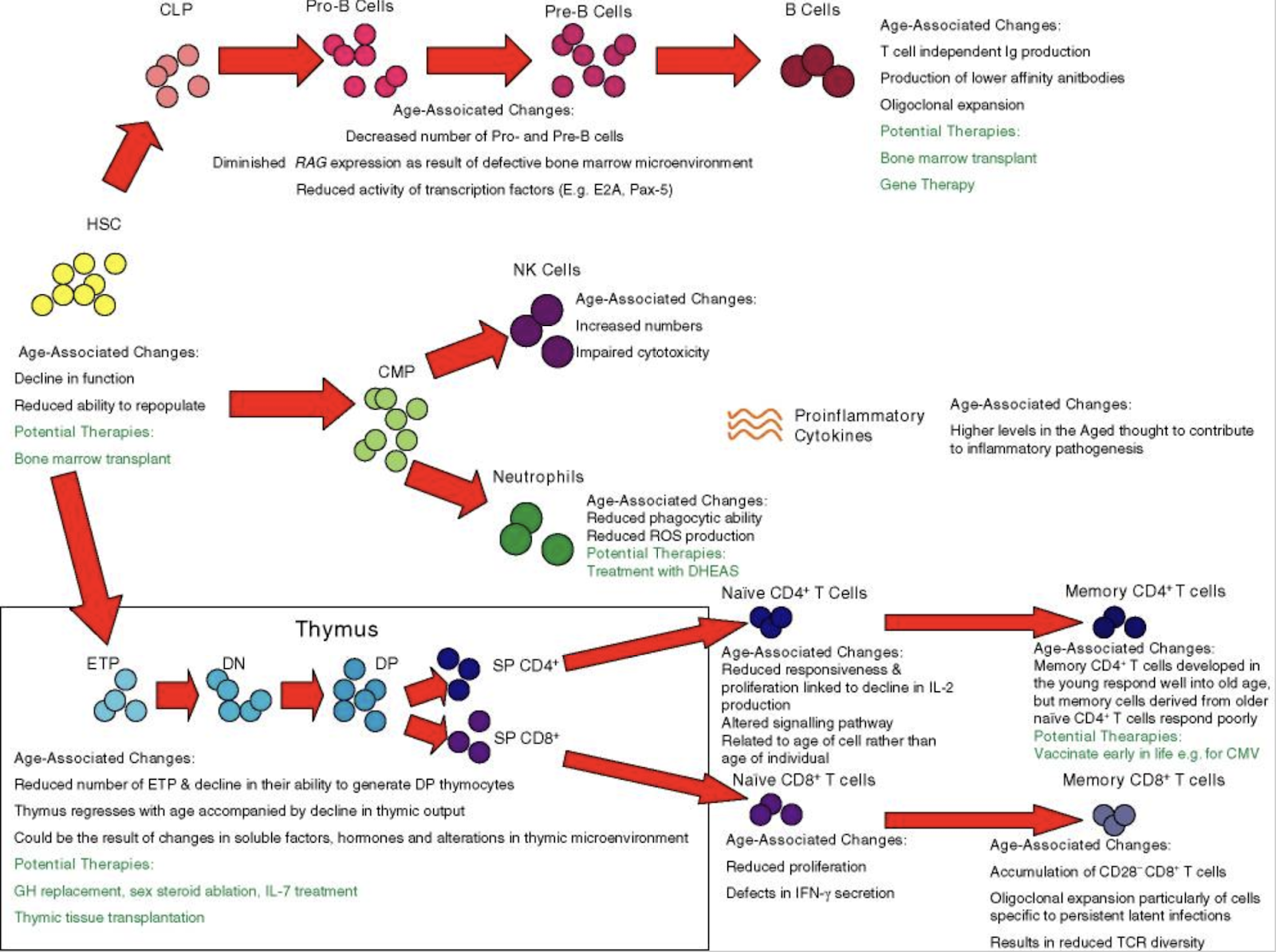
Impaired response to vaccines (and infectious disease)
Fortunately there is no need to write a section here! Brandon White has already compiled a great source convering this. The review goes more in depth into non-T cell aging than my own article.
Thymic involution
The thymus is a gland located in between the lungs, in our chest. When we are born the gland is maximally active and trains T cells to recognize antigens and not attack the host. As we grow, it undergoes involution at an exponential pace. It doesn’t fully go away, but its size and function shrink until it is little more than a lump of fat. This has grave consequences: Without the thymus, T cells are not properly trained to avoid attacking the host, increasing the odds of autoimmune diseases, and naive T cell production ceases, as that requires maturation in the thymus.
Thymic involution doesn’t happen at random; it is a regulated event and while the full regulatory network remains unknown, a few proteins have been found to be implied in the involution process.
Most work I found universally refers to FOXN1 as the master regulator of the involution (Chen et al. (2009), Vaidya et al. (2016)), and its levels predictably decrease with age; for example FOXN1-deficient mutants that show a faster rate of decrease of FOXN1 after birth also show faster atrophy as measured by thymocyte numbers.
Upstream of FOXN1 the full picture is not fully formed yet, with one lead candidate being the E2F family of transcription factors, the Rb1 (retinoblastoma tumor suppresor) gene (Song et al., 2017), which suppresses E2F activity (Garfin et al., 2013), PPAR-gamma (Ernszt et al., 2017), Wnt4 (Wei et al., 2015) and TBX-1 (Kvell, 2019). TBX-1 is an interesting case because its absence is what leads to DiGeorge syndrome. However its presence during adulthood acts as a promoter of the conversion of the thymus into adipose tissue. Micro RNAs are also involved in the process, but how exactly is still not fully understood (Xu et al., 2017, Bernardi Bertonha 2020). This is my tentative diagram of the key TFs (dots mean downregulation):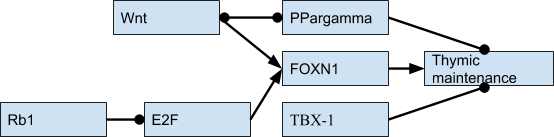
There are other factors that are also involved, namely sex hormones and growth hormone, however their effects seem smaller than those of the regulated FOXN1 program. For example, castrating mice does lead to an increase in volume of the thymus, but it eventually shrinks again.
The involution is not complete: Following hematopoietic stem cell transplantation, which involves whole body irradiation or chemotherapy, destroying the entire immune system, what is left of the thymus is still able to steadily recovery naive cell counts and TCR diversity (van den Broek, 2018, Politikos & Bou, 2014) over a long time; this process being faster in the young (Hakim et al., 2005).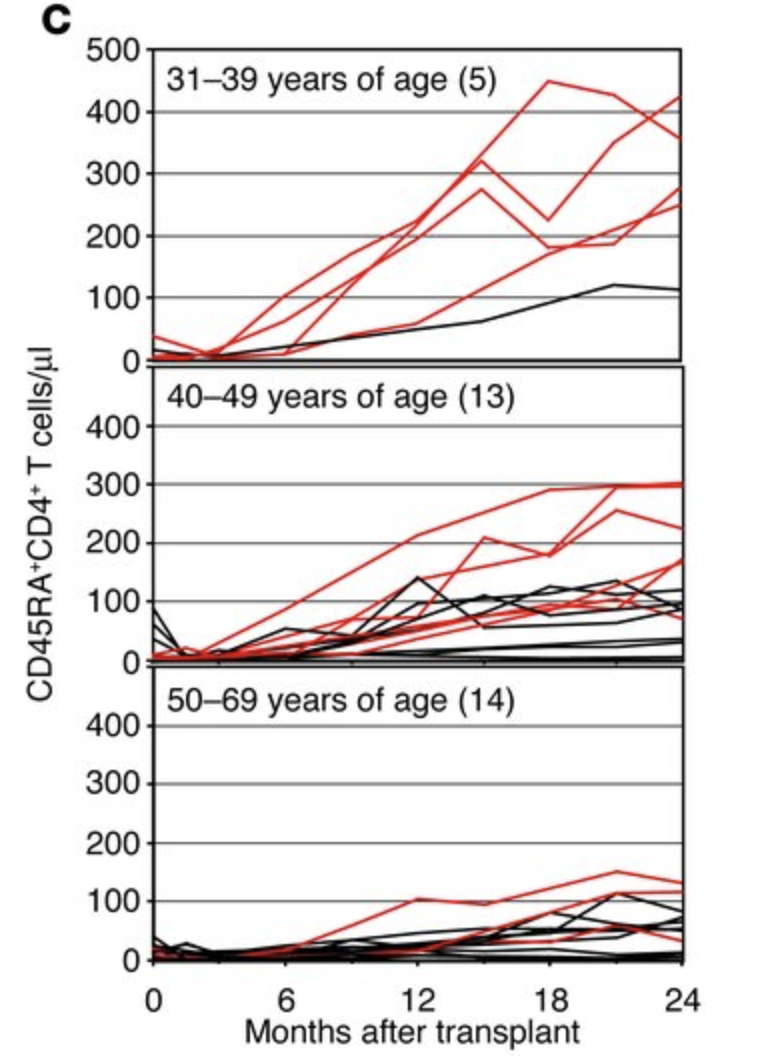
How fast does the involution occur?
Thymic involution is best modeled as an exponential decay process, and so it is worth looking at half-lives: How long does it take for the thymus to reach half its original size or output.
den Braber et al., 2012 give a rate of thymocyte production decay of 50% a year, or a half life of 365 days for C57BL/6 mice.
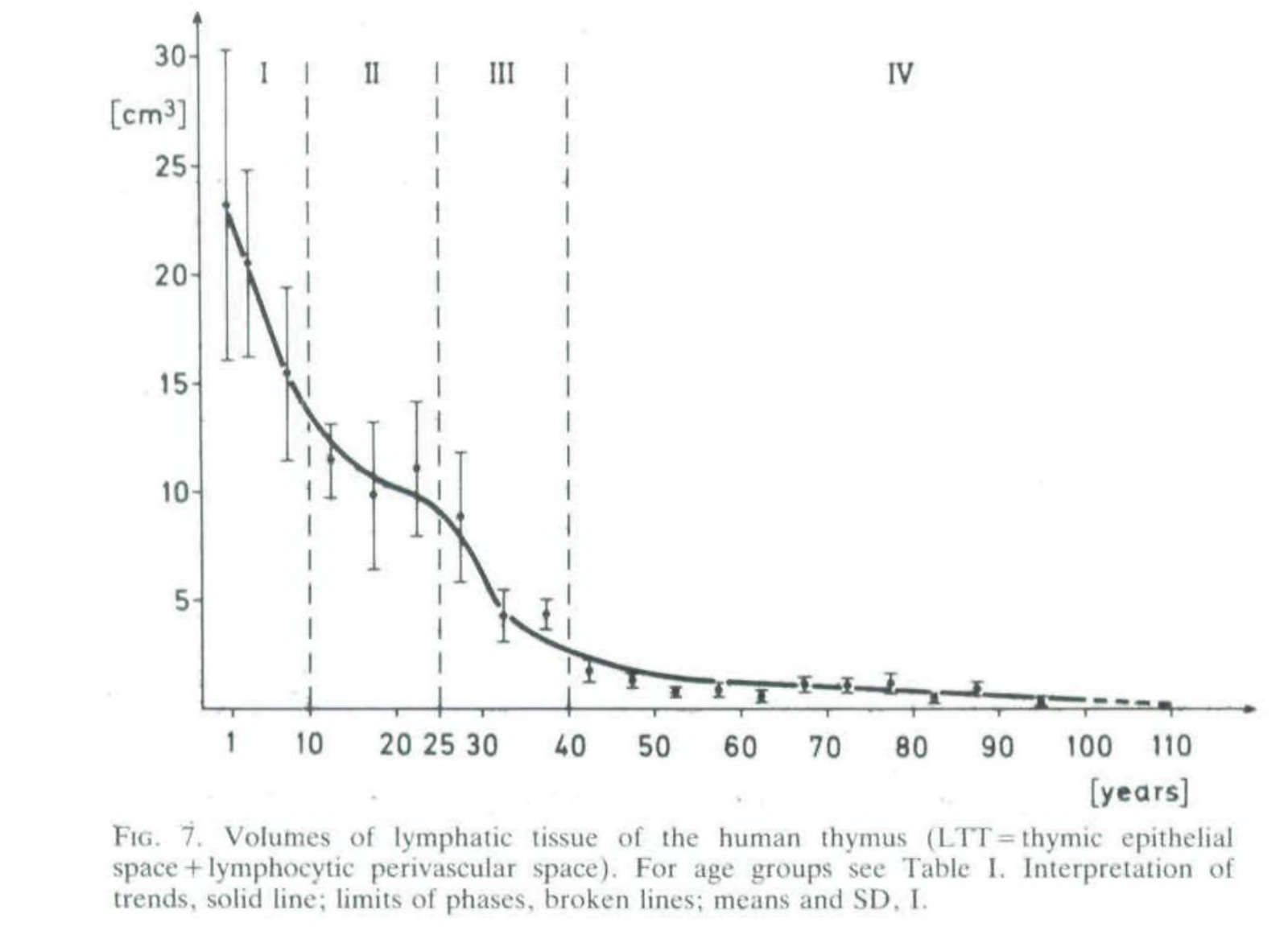
In humans the important parts (medulla, cortex, and lymphocytic perivascular space) of the organ itself have a half life by mass of 23 years initially, possibly slowing down to 67 in the old age (from decay rates in Steinman et al. 1985) and half-life for thymocyte decrease is 15.7 years (Palmer 2018) 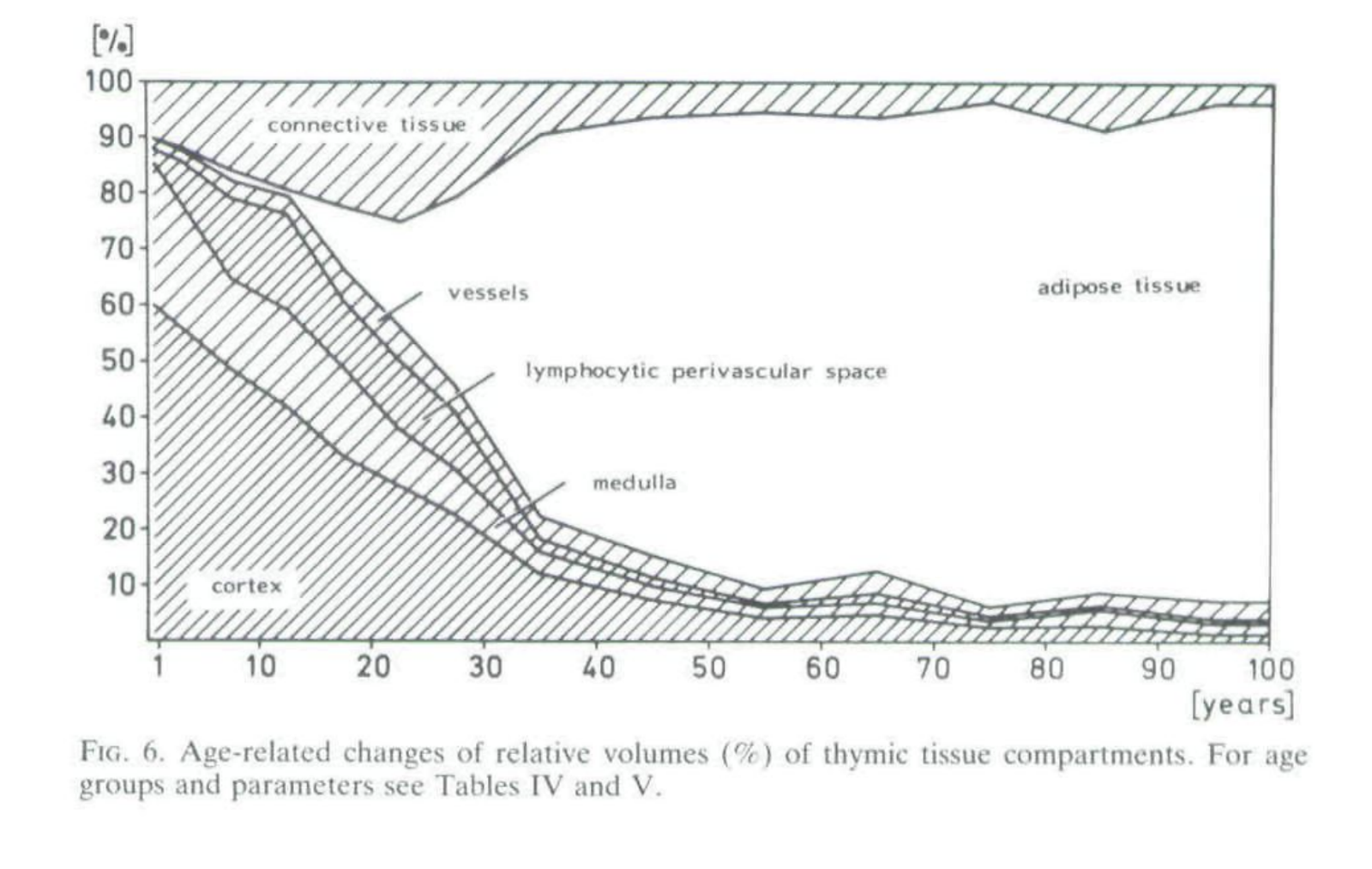
 Mice vs humans: One important difference between thymic activity in humans and mice is that in mice most of the circulating T cells are coming from the thymus, while in humans in adulthood (90%) or later (99%) most T cells come from clonal expansion: T cells in mice almost don’t divide (den Braber et al., 2012, van den Broek. 2018). This means that studies looking at T cell senescence or TCR diversity or naive T cell populations will be biased in mice studies, making it seem like it is not that bad compared to humans. In humans we are stuck with a shrinking pool of naive diverse T cells, mice are constantly refreshing theirs at rates two or three orders of magnitude greater than us,
Mice vs humans: One important difference between thymic activity in humans and mice is that in mice most of the circulating T cells are coming from the thymus, while in humans in adulthood (90%) or later (99%) most T cells come from clonal expansion: T cells in mice almost don’t divide (den Braber et al., 2012, van den Broek. 2018). This means that studies looking at T cell senescence or TCR diversity or naive T cell populations will be biased in mice studies, making it seem like it is not that bad compared to humans. In humans we are stuck with a shrinking pool of naive diverse T cells, mice are constantly refreshing theirs at rates two or three orders of magnitude greater than us,
T cell reconstitution in adult humans is more comparable to that in thymectomyzed mice than that in euthymic mice (Mackall and Gress, 1997). It is important to realize that these quantitative differences between mouse and man will also have qualitative effects on the naive T cell pool because the thymus is capable of producing new T cell specificities, whereas peripheral T cell proliferation can only lead to the expansion of already existing T cell clones.
Does thymic involution depend on age?
Suppose an old mouse gets a new thymus from a newborn. What happens to that thymus? Will it immediately collapse to the shrinked size of the original thymus, driven by external factors to adopt an aged configuration, or will it instead steadily decline in the same way as the original one? In a surprising study where they grafted up to 48 thymic glands (yes, 48, don’t ask, you can read Metcalf 1965 directly) in mice aged 2 and 20 months each small implanted thymus actually grows to around its full size almost as well in both groups of mice, showing that the involution seems to be driven by factors internal to each thymus, and not so much by, say, factors in the blood.
More recently, Zhu et al. (2007) found that 7 weeks after grafting fetal thymuses to young(2m) and old (20m.) mice the grafts in both mice were equally active by total thymocyte count and by T cell population types.
So it seems that no, the process is independent of age or anything external to the thymus.
Does fixing thymic involution help with lifespan?
Hirokawa et al. (1984) took adult mice (15 in each group) and serially transplanted thymi into the peritoneal cavity, one every month for 4-5 months. This didn’t extend maximum lifespan, but extended median lifespan by 46% from the start of the treatment, relative to control mice, however the authors believe that continuing the protocol would lead to an increase in maximum lifespan.
While done in nude (athymic) mice, Basso et al. (2010) showed that transplants from newborn mice to old (114 weeks) mice (52 mice per group) increased maximum lifespan by 10%, with a similar change in median lifespan, so small Kaplan-Meier analysis yield nonsignificant differences; the authors preferred to use a more sensitive parametric model to support their conclusions. Being skeptical, this is also compatible with a null result.
Kulikov et al. (2014), by means of grafting thymic tissue into the eyes(!) of adult (16 months) rats (24 control, 21 treatment), showing a 20% increase in medium and maximum lifespan. As the rats presumably would have trouble seeing, maybe there were other effects; if they were eating less that would have increased lifespan too.
More indirectly we can look at thymic function and mortality, for a given age does it help to have a more active thymus in humans?
Derhovanessian et al. (2013) paradoxically found that in a 85 years old cohort fewer naive cells predicted lower all-cause mortality, they interpret this result as meaning that having more differentiated memory T cells present allows a better response to the antigens that population would be regularly exposed to; naive T cells would come handy in the less frequent case they are exposed to new pathogens or cancer. Not everyone buys this straightaway: Weltverde et al. (2016) ask for more research before concluding that high naive T cell count is a negative in the elderly population. In particular they complain about the lack of standardized markers to define different T cell populations, which makes it hard to compare studies. Also this finding was not replicated in an age-stratified sample of patients with renal disease, naive T-cell count negatively correlated with all cause mortality at 35 months (Zhou et al. 2020). Likewise in a cohort with a wider span (65 to 102 years) thymic function (so measuring “new” naive T cells) predicted all-cause mortality (Higher count of naive T cells was associated with survival); for that matter also did neutrophilia and an inverted CD4 helper/CD8 cytotoxic T-cell ratio (The Immune Risk Phenotype, IRP) (Ferrando-Martinez et al. 2011)
TCR repertoires and naive cells, a red herring?
Naive cells are produced exclusively in the thymus and are also the most TCR-diverse population there is (van den Broek et al., 2018). T cells can be grouped in clonotypes, a group of T cells, each with their TCR receptor that react to the same antigen. TCR diversity in blood is 3x lower in their long lived(>92) sample than what is found in umbilical chord blood and 2x lower in the aged (51-75) sample, while naive T cells decline from 80% of all T cells to just 20% (Britanova et al., 2016). Women have consistently higher TCR diversity and naive T cell % compared to men regardless of age except perhaps in the very elderly. This diversity is also unequally distributed: On any given sample they found 0.4-0.6 million of distinct TCR receptors, but between 45 and 75% of all T cells belong to the group of the 1000 most frequent. In reality this will be worse because as I mentioned earlier, blood measurements underestimate the effects degree of involution in naive cell counts.
Is a reduction in TCR diversity really a problem? The reduction is there but quantitatively there is still a lot of it; in particular Goronzy & Weyand (2019) and Jergovic et al. (2018) are of this view. The latter even question that even the combination of reduction of naive cells and TCR diversity are enough to account for the weakening of the immune system: They even question that the cells themselves are somehow impaired:
in vitro studies have shown CD8+ T cell intrinsic alterations with aging on an epigenetic level, however research showing functional defects of aged CD8+ T cells is confounded by different subset representation, and no significant defects on a per cell basis have been demonstrated. [,,,] Our recent transfers of purified naïve CD8+ T cells (Fig. 1.) isolated from aged TCR transgenic mice have repeatedly shown no intrinsic functional decline when these cells were primed in the adult recipient.
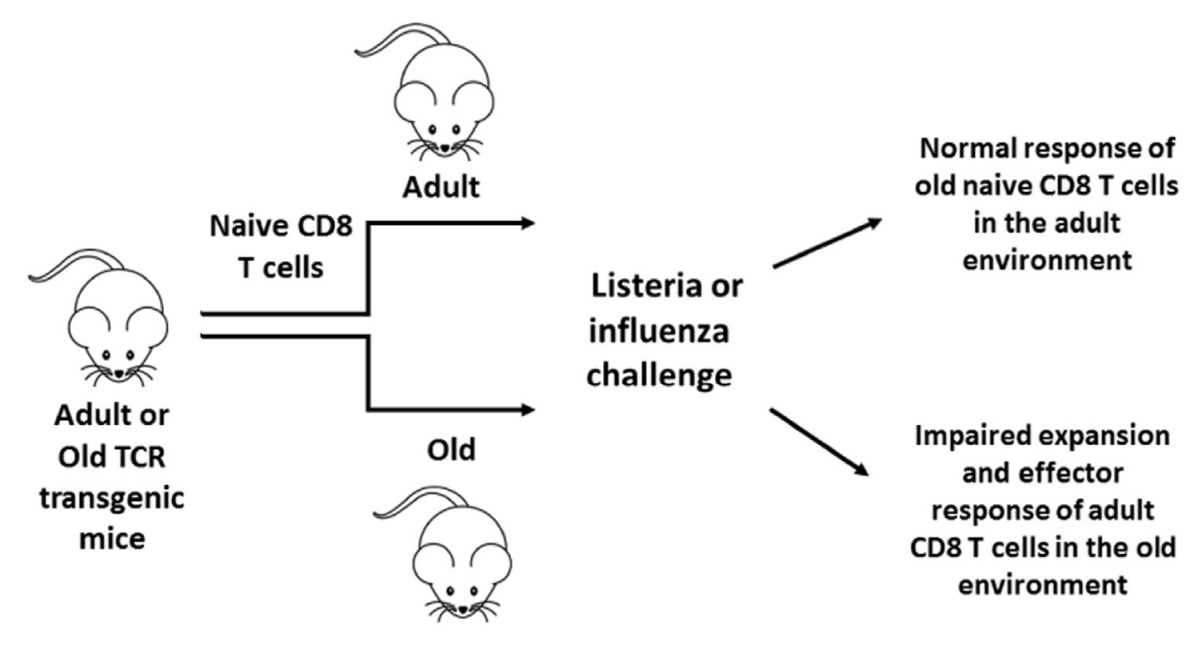 So it’s that they don’t get properly activated then. Ok, but why? It is the activation of the immune response that is to blame, they say. The ones that get it going are macrophages, B cells, and antigen-presenting cells like dendritic cells that go show them to the T cells. Only dendritic cells (DCs) can activate naive cells (Kambayashi & Laufer, 2014) so maybe those are to blame?
So it’s that they don’t get properly activated then. Ok, but why? It is the activation of the immune response that is to blame, they say. The ones that get it going are macrophages, B cells, and antigen-presenting cells like dendritic cells that go show them to the T cells. Only dendritic cells (DCs) can activate naive cells (Kambayashi & Laufer, 2014) so maybe those are to blame?
Studying the entire response process to pathogens at the cellular level has proven hard: It’s not just that you need the T cells and the dendritic cells, but also other cells that also interact with DCs like neutrophils or NKs. So Jergovic et al. (2018) point to in vivo studies in mice where they try transferring bone marrow with or without DCs into old mice (or adult vs young DCs), finding that indeed it is the DCs in particular, and not so other cells in the bone marrow that are doing the trick (Stout-Delgado et al., 2008) . So if DCs are a key player here, the authors expect that rejuvenating secondary lymphoid organs (where T cells meet DCs) and restoring intercellular communication would be a highly impactful intervention. To be sure this is an hypothesis, albeit one that the authors think would suppose a “Copernican revolution” in immunosenescence if accepted, as it shifts the focus of the field away from where most of it seems to be right now. For now, most of the reviews still seem to say that DC aging is still poorly understood (Pinke et al., 2018)
Impact of cytomegalovirus
Cytomegalovirus (CMV) is a virus with a surprisingly high prevalence: Zuhair et al. (2019) estimate that a large fraction of the world’s population is infected by it, with extremes being Turkey (almost 100%) and Ireland (closer to 40%) with the US around 70%, and this number increase with the age of the subjec getting closer 10 100% in the US in those over 75% (Dowd et al., 2008). The virus itself is remarkable: Its genome is quite large (240kb, see Stern-Ginossar et al., 2012), perhaps 20x as large as the flu virus and like other herpesviruses possesses a remarkable ability to hide in the body, fooling the immune system for decades, and periodically reactivating itself, producing generally no symptoms.
But just because there are no symptoms doesn’t mean the virus is harmless. With a declining pool of naive T cells, constant reactivation of CMV has been proposed as the cause of “memory inflation”; or a crowding out of other TCR clonotypes by the CMV ones which eventually contribute to weakening the immune response to more harmful threats. Paradoxically, in young individuals both human and mice, CMV (the human and mice varieties, respectively) seems to strengthen the immune system presumably by raising the state of alert of the immune system. CMV generally an issue in immunosuppressed individuals, such as those undergoing chemotherapy or those taking immunosuppressant drugs, like high doses of rapamycin (Jergovic et al., 2019). I however will leave these cases aside and focus on its effects in the old age.
Memory inflation in practice means that on average 10% and up to 50% in some cases of all memory T cells are CMV-specific. This has been linked to an increase in total circulating memory T cells that wouldn’t be otherwise observed in CMV-free individuals, but due to the low antigenic load the virus supposes this has not been linked to an increase in replicative senescence of T cells, rather the bulk of the cells dedicated to CMV surveillance belong to the so called Temra (T effector/memory cells that express CD45RA) subpopulation which do not replicate, but unlike senescent cells don’t have a SASP or are otherwise impaired.
All things considered, what role does CMV play in immunosenescence?
Weltvrede et al. (2016) in a systematic review that claims to be the first one examining the relation between immunosenescence and CMV in individuals > 50, report the intriguing finding that despite the fact that most studies report a shrinked naive T cell compartment in CMV-positive individuals, high quality studies find no such effect. What the studies reviewed did find uniformly was an increase in memory and CD8/4 Temra cells and that CMV leads to higher counts of CD4/8 T cells, to some extent explained by those memory cells. As for the CD4/CD8 ratio, it remains unclear if the CMV is causally associated with it, and the authors are skeptical of using the ratio in clinical applications due to insufficient validation
Jergovic et al., 2019 claims that it is not really a factor:
Overall, while CMV has a massive impact on the T cell compartment and the immune system regardless of age, the initial hypotheses reviewed in [42, 77] that CMV may drive immune senescence, loss of TCR diversity and, directly, or indirectly, reduced responsiveness to third-party antigens/ infections have either not been substantiated in critical, incisive studies, or have been rendered less likely by the available evidence.
In this video, Graham Pawelec (one of the most cited experts in this space) points to evidence that CMV infection reduces the number of circulating naive T cells (I couldn’t find the exact forthcoming Goldeck paper he refers to, but Goldeck also coauthored Di Benedetto. 2015 and makes the same point with the same data). Figures c and d on the paper are not very clear about whether or not there are differences among the old infected or not with CMV and I wouldn’t take the authors’s conclusion that there is a difference, but only for women, as strong evidence for that claim.
CMV doesn’t appear to reduce TCR diversity either (Lindau et al., 2019): memory inflation is real but more of a one-off event, a fixed pool of CMV-dedicated T cells suffices to keep the virus under control, with no ever increasing accumulation of these cells occurring over time.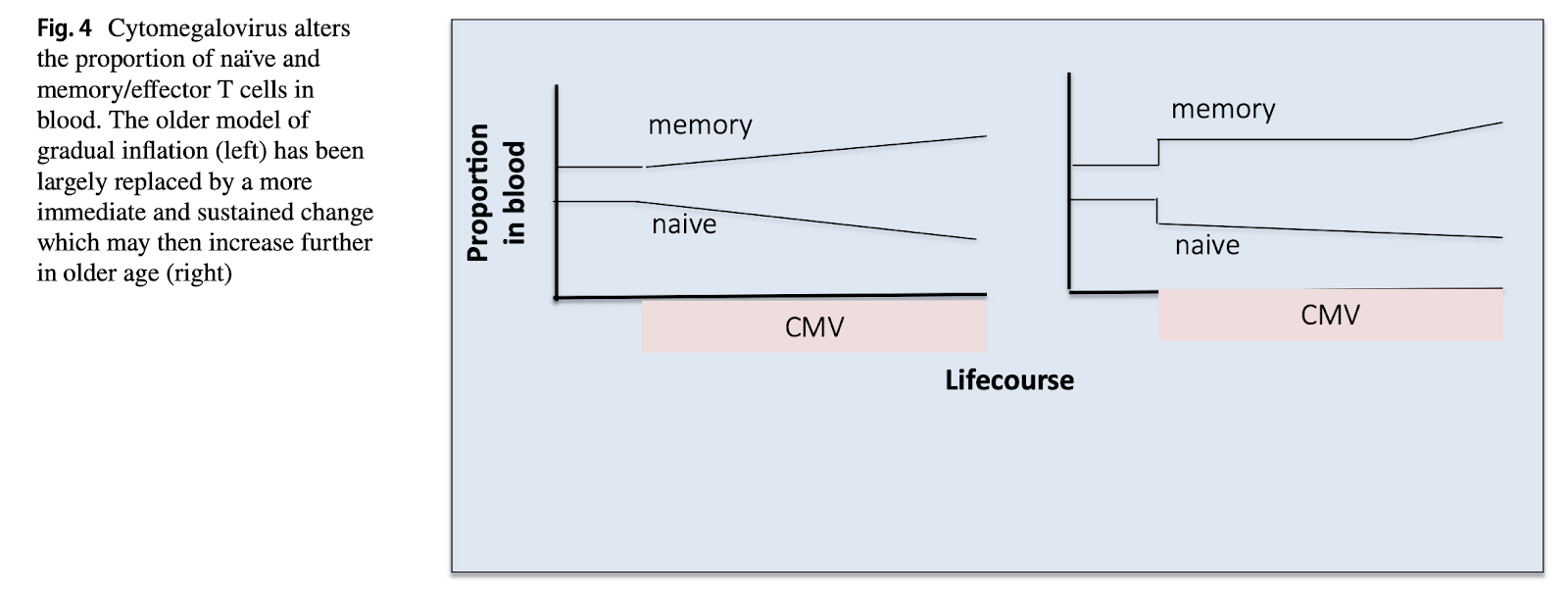
Lastly, two years ago there was the 6th International Workshop on CMV and Immunosenescence (Nikolich-Zugich & van Lier, 2017). An event that brings together the experts in the field is always worth reading the proceedings of, and as is in agreement with the above, serves as a conclusion here:
- It is not yet known for sure if CMV negatively affects responses to other pathogens
- CMV infection does not cause a reduction in the naive T cell compartment
- The contribution of CMV to immunosenescence is a topic that continues to elicit debate among experts, as a core set of evidence, large epidemiological studies do not give clear answers
- CMV affects mainly T cells seen in blood and the spleen, not so tissue-resident T cells (also see Moss, 2019)
The paper ends with a series of recommendations for CMV researchers that include: stop sampling just blood, stop assuming that CMV-reactive T cells are exhausted, standardise methods, and getting a clearer view of the viral activity by means of high-throughput sequencing of the transcriptome of the virus itself and cells infected by it. I also have to note that in the same issue of that journal, Aiello et al. (2017) also share evidence that associated CMV infection with everything from atherosclerosis to cancer and diabetes, though they admit that the causal role of CMV is debatable, and I add, possibly confounded by other factors like socioeconomic status, which is also associated with CMV status and those illnesses.
All things considered this seems an area that is still not settled. Pawelec, in the video linked, seems more of the opinion that CMV effects are real but highly context dependent based on gender or population studied, but no true randomized studies have been done, it’s hard to know what exactly CMV is causing, other than proliferation of Tem/Temra cells dedicated to combat it. My overall view is that CMV is probably not a key driver of immunosenescence.
Stem cell depletion and microenvironment changes
HSCs coexist with others like macrophages, mesenchymal stem cells (MSCs), adypocites, or osteoblasts, with these cells being fundamental for HSC expansion and maintenance (Latchney & Calvi, 2018). Collectively this is referred to as the microenvironment, which is also hypoxic in nature (promotes quiescence and glycolisis, which generates less ROS). Bone marrow is also innervated, thus bringing the brain as one more potential factor in HSC aging (Maryanovich et al., 2019, Ho et al., 2019), with the degree of innervation and signals changing with aging.The raw material for functional, healthy, white blood cells are HSCs that reside in the bone marrow. Some authors say that these cells multiply their numbers by a factor of 2-10x through life, which seems paradoxical to say in a section titled “stem cell depletion”, but this increase in numbers may be related to loss of functionality: maybe defects in the cells lead them to multiply more. The “hallmarks” of HSC aging are (Lee et al. (2019) Buisman et al. (2019), Latchney & Calvi, 2018, Pinho & Frenette, 2019):
- There are conflicting reports about whether HSC counts increase (Farrell et al., 2014) or decrease with age, as noted for example by Thomas et al., (2020)
- Decreased homing ability (to find their way to the right place in the bone marrow)
- Skew towards the myeloid lineage (universally agreed)
- Decreased diversity, most of the extant cells in the elderly are derived from a progressively smaller pool of cells, only the “fittest” survive and get to reproduce (Busque et al., 2018)
- Reduced proliferative ability (disputed)
- Replacement of hematopoietic marrow by fat starting around adolescence (Pinho & Frenette, 2019)
While it used to be believed that the above were to some extent due to DNA mutations, the consensus now seems to tie them more to epigenetic degradation and damaged microenvironment (Moerhle & Geiger, 2016, Guidi & Geiger, 2017). These epigenetic changes (in the histones, increased methylation of sites related to genes that promote differentiation and the myeloid skew). are not reverted when transplanted so they appear to be not a response to external conditions, but a causal factor of the aged phenotype. This needs more investigation.
In addition, the microenvironment matters too: as in the thymus, the bone marrow becomes increasingly fatty with age (Tuljapurkar et al., 2011), reaching up to 80% in some bones in women (Veldhuis-Vlug & Rosen, 2017) with an alteration of various chemokines present in the healthy marrow which play a role in the bias towards the myeloid lineage (Wang et al., 2018).
In turn, why does the bone marrow turn into fat? Lack of oestrogen is one factor that accelerates the process after menopause in women. The parathyroid hormone (PTH), which plays a role regulating calcium, has been shown in mice and rats to reduce fattification when administered periodically. In humans however this hormone goes up with age (Carrivick et al., 2015), perhaps trying to counteract some other tendency? In any case, I leave this topic outside of the scope of this review.
Overall this literature seems in flux and there doesn’t seem to be agreement about basic facts like whether or not HSCs increase in number in humans, or to what extent are HSCs functionally impaired with age. About this latter, the direction of the field seems to be in the direction of HSCs being mostly fine, and the culprit being the microenvironment. Some studies below:
Zhu et al. (2007) finds that early T cell progenitors (ETP, that come from HSCs) from young mice are not able to recover immune function in old mice, but the opposite is true: When implanting old ETPs into young mice with a defect in the medulla of the thymus, that does recover thymic function., meaning that the cells are not damaged in any sense, they just lack the right microenvironment The author also mention that other studies are confounded by the fact that HSCs tend to be more committed to the myeloid rather than the lymphoid lineage with age, so what ends up happening is that wholesale transplant of HSCs won’t pick up as many lymphoid-committed cells that are the relevant ones for the thymus. Here, the authors conclude that efforts to rejuvenate the thymus should focus on the thymus itself, not the thymocytes. (Except perhaps for the very aged, as noted by Lee et al. (2019) ). Ironically, this finding itself may be due to selection bias: It may be that cells that manage to become ETPs are in a better condition than the others. 
However, that doesn’t mean one can’t target the HSC microenvironment to avoid them being biased towards becoming myeloid cells. Young et al. (2020) study what is causing this bias, finding that it is, among others decline in IGF1 in the bone marrow. Ex vivo supplementation with IGF1 didn’t fully restore the commitment % of HSCs, so there’s more to this. Deleting the receptor of IGF1, IGF1R increased commitment to the myeloid lineage, so the mechanism seems robust.
This is somewhat surprising because the basic story with IGF1 is that lowers levels of it correlate with longevity. But the findings are not at odds: It can be that higher chronic circulating IGF1 is bad, but pulsed IGF1 is good, in coherence with the fact that growth hormone expression occurs in pulses through the day, the amplitude of which declines over time (Garcia et al. 2019). In mice, administering low-dose GH twice a week to mice (17 months old) result in greatly reduced mortality, with 61% of the control group dying vs 7% of the treated group over a given time period. In fact, median lifespan of the treated group was - greater - than the maximum lifespan of the control group: By the time the control group had died, half of the treated group was still around (Khansari & Gustad, 1991). This didn’t replicate in rats though, where no negative or positive effects were seen for that specific dose (Kalu 1997). One possibility is due to better housing conditions in the Kulu case (fewer infections in the control group), but again worth investigating.
Macroph-aging
As mentioned earlier, macrophages phagocyte pathogens, present antigens to T cells, and secrete cytokines to regulate the immune system as well. There are various kinds of macrophages: By function authors tend to distinguish between M1 (pro-inflammatory and glycolisis powered) and M2 (anti-inflammatory and OXPHOS powered). Macrophages, given the right stimulus, can switch from M2 to M1, but rarely the opposite (Italiani & Boraschi, 2014). (This is a simplification as M1/M2 is more of a spectrum)
Not all macrophages are created equal: At birth we are equipped with a set of tissue-specific macrophages that self-renew, and these stick around for life. In parallel, the bone marrow produces other macrophages that tend to be in blood or lymph node, or exceptionally migrate to tissues. In adult mice, macrophages in the pancreas, liver, brain, or the skin are mostly derived from the originary fetal-derived population, while the rest gets slowly replaced with HSC-derived ones (Gomez Perdiguero et al., 2015, Roszer, 2018)
Macrophages would typically be going around phagocyting stuff but they have recently been implicated in another correlate of aging: NAD depletion. Turns out M1 macrophages express the enzyme CD38 (Hogan et al., 2019), a NADase that makes NAD into nicotinamide (NAM). Oddly this enzyme has its reaction side outside of the cell, so it dumps NAM on the outside. One hypothesis for the function of CD38 is that this is because NAM may have anti-inflammatory properties, or that depriving pathogens of NAD is good to prevent their replication. However, when faced with an increased number of senescent cells (Chini et al., 2019), they upregulate CD38 (600x over their baseline per Covarrubias et al., 2019, which also show through a large battery of assays that it is indeed CD38 in M1s and not PARPs or sirtuins that are to be blamed here) which causes NAD levels to plummet. This action is particularly notorious in adipose tissue and the liver, relevant as the liver is 15% made up of macrophages and 80-90% of all tissue-resident macrophages are in the liver.
Naturally, this raises the question of whether getting rid of senescent cells reduces the action of these NAD-thirsty macrophages. It would be very convenient if a) Senescent cell clearance reduces NAD depletion and b) A rejuvenated immune system can actually clear those senescent cells.
But that’s just regular macrophages faced with odd circumstances (too many senescent cells). Is it the case that the macrophages themselves change as we age?
Van Beek et al. (2019) note that a lot of the studies done on macrophages are ex vivo, and from there misleading results may follow: macrophages are not all the same, and their behaviour changes depending on tissue-specific cues that may be absent ex vivo. For example, macrophages in alveoli and peritoneum of mice express a different set of genes. But when you take an alveolar macrophage and put it in the peritoneum, it gets reprogrammed to become an alveolar macrophage. That said, and looking at concrete intrinsic changes, it is unclear if their phagocytic activity is impaired with aging. Instead they side more with the idea that it is exogenous changes: deterioration of the gut epithelium lead to bacteria leaking out, which trigger inflammation; so does the accumulation of AGEs. Why does DHEA decrease? Because the adrenal glands that make it involute( Samaras et al., 2013).
Their polarization might depend on the presence of senescent cells. In the muscle or the lung there are almost no senescent cells, while they substantially increase in the spleen, kidney, skin, liver, or the brain (Idda et al., 2020), perhaps explaining why in muscles macrophages are M2s, albeit perhaps odd pro-inflammatory ones (Cui et al., 2019) while Covarrubias et a. (2019) found lots of M1s in adipose tissue instead.
As for intrinsic changes, there are there too: They take longer to react to LPS or they tend to prefer the M1 inflammatory/glycolytic polarization. Mitochondrial dysfunction forces cells to use glycolysis which in macrophages skews them towards the M1 type. Why would they accumulate damage? Macrophages are regularly turned over in the order of days (Patel et al., 2017) or weeks (Ginhoux & Guilliams, 2016) but there are reports in mice where tissue-resident macrophages in the lung (Murphy et al. 2008) are not replaced and live as long as the animal lives. Maybe there is more room to accumulate damage in these cells, which unlike HSCs are not shielded inside the bone marrow.
Van Beek et al. conclude that maybe something that could reconciliate the disparate findings is to take into account the fact that the innate immune system is somewhat adaptive, macrophages have some memory:
In view of the currently available experimental evidence, an attractive scenario describing the mechanism underlying alterations at the macrophage level during aging might involve the phenomenon of innate memory. This has been described recently as trained immunity, explaining enhanced responses, but also includes the longer known induction of tolerance [81,82]. Chronic exposure of long-lived tissue macrophages and bone marrow precursors to increasing levels of inflammatory triggers, such as LPS, S100A8/A9, and products from dying or senescent cells, might impose epigenetic changes that cause altered responsiveness of macrophages during aging. Together with intrinsic changes induced by a decreased ability to repair cellular damage, this might lead to an enhanced, or sometimes decreased, response to an acute trigger, such as that occurring from infection or metabolic perturbation.
Senescent cell clearance
The immune system clears senescent cells (Kale et al., 2020, Ovadya et al., 2018), and there are proposals of using CAR-T or CAR-NK cells can clear senescent cells (Amor et al., 2020). A lot of the T cells in the elderly are actually senescent, up to 64% for CD8s (Martinez-Zamudio et al., 2020), so plausibly clearing senescent cells in general might be useful for the immune system as well, although if too high a dose were to be administered perhaps too many T cells would be lost; which would be replenished mostly by clonal expansion, making it worse.
But what is of additional interest here is knowing if either thymic rejuvenation or some similar treatment also reduces senescent cell count (Other than senescent T cells). That has not been studied yet, but it stands to reason that they should have an effect. If confirmed, a single intervention could, in the most optimistic case, do what is currently being attempted with NAD boosters and senolytics, combined.
Natural Killer Cells
NKs are lymphocytes that belong to the innate immune system; they do not mature in the thymus, and canonically defend against cancer or infected cells but are also implicated in clearing senescent cells or modulating inflammation (A general theme in the immune system: cells are vastly more multi-functional than originally thought). NKs do the latter by, among others, directly attacking and removing DCs, neutrophils, or T cells. The rate of production of new NKs decrease with age, but the total number increases, reflecting an accumulation of long-lived NKs. Whether their cytotoxicity or capacity to migrate to areas where they are required are impaired with age is unclear (Hazeldine & Lord, 2013). Some, like Guo et al. (2014) think it may be dendritic cells all along: NKs from old mice behave almost like young NKs when stimulated by young dendritic cells in vitro. In vivo, when ablating DCs from young mice and injecting them DCs from old (18mo) and young (3mo) mice, injected cancerous cells were more often eliminated in the later (2.5% remaining) than in the former case (15% remaining) after 12-14h. However, repeating the experiment with human NKs and DCs the results were not as clear; the authors claim that maybe their “old” sample (55-70) years old is not that old for the effect to show up, which to me seems a bit odd.
Regulatory T cells
Regulatory T cells (Tregs), like other T cells are trained in the thymus (plus they can arise from ciruclating naive T cells); and their role is to downregulate proliferation and action of T cells or ultimately attack them and kill them with granzyme B. As with elsewhere in this review, whether a T cell is a Treg or not seems to some extent to be context dependent: With the right stimuli Tregs can become effector T cells (or viceversa). There are Tregs both inside the CD4 and the CD8 T cell populations.
With thymic involution, total lymphocyte production goes down, but for effector T cells, they become less able to recognize the own body, what could lead to potential autoimmune disease. At the same time, an increase in Tregs is observed. While this is an appropriate response to contain defective effector T cells, too many T regs can make the immune system too conservative against infections or cancer. While this story is true in a broad aggregate sense, it is not when disagregating by types of cancer, in particular it is not true for colorectal, head and neck and oesophageal cancers where more Tregs infiltrating the tumor correlate with better survival; perhaps because the suppression of pro-inflammatory innate immune system cells (which are more present in those cancers) outweighs the suppression of T cells (Shang et al., 2015, Sakaguchi e tal. 2020).
Back a few years ago it was unclear whether Tregs increased or not with aging, with reports pointing in contradictory directions (Raynor et al., 2012). Now we know that there are diverse Treg subpopulations and some increase with age, others decrease, and for others we still don't know; while in the aggregate they seem to increase (Jagger et al., 2014). In the case of humans, Tregs of thymic origin increase, and "induced" Tregs (originary from circulating ) T cells decrease. In the aggregate, the pool of Tregs in the elderly is not as capable as the young ones, their ability to suppress self-reactive T cells is impaired.
it is unclear where the increase of aggregate T regs is coming from, but Jagger et al. discuss various hypothesis, ultimately settling on the fact that Tregs become better at surviving than CD4 effector cells, so the former expand at the expense of the latter. CD8 Tregs appear to follow the same patterns.
Cancer
The standard story with cancer is that mutations accumulate at random and then eventually a combination is reached that enables uncontrolled cellular growth. Sometimes this will happen earlier if one has been born with that mutation or later if not. Also some cancers will affect children more than adults (Like retinoblastoma) but those cancers don’t overlap much with the cancers of adulthood; for example there are literally only a handful of prostate cancer cases in children; in the period 1935-1949 out of 2532 prostate cancer cases, only 2 were in children (Griswold, 1952), or between 1974 and 2007, there are only reports of 11 cases of colorectal cancer in children (Salas-Valverde et al., 2009). Conversely, there are only 45 cases ever recorded of retinoblastoma in adults (Sengupta et al., 2016). Heart cancer is rare even in adults (Sarjeant et al., 2003). Naively one would expect that if cancer is just the unavoidable outcome of rare mutation events, then the more cells there are in an organ the higher the rate of cancer there, but this wouldn’t explain why prostate (a walnut sized gland) cancer is so prevalent while heart cancer (more of a fist-sized organ) is so rare.
It is not yet known why different organs are differentially afflicted by cancer, some speculation points to more essential and less exposed to the outside organs are more protected (Thomas et al., 2016).
It may seem intuitive to think that larger organs should have higher cancer rates, but no, and this result is echoed across animals (Tollis et al., 2017): blue whales don’t have higher cancer rates than humans despite there being orders of magnitude more massive, having evolved additional anti-cancer mechanisms. As for how many mutations are needed for cancer to happen, it is a surprisingly low number, between 2 and 8 depending on the organ (Anandakrishnan et al., 2019), with the top suspect being mutations in the p53 gene that regulates apoptosis (Mantovani et al., 2018).
Besides mutations, the decline in immune function has also been postulated as a contributor, perhaps the main contributor, to the rise in cancer risk through age by Palmer et al. (2018), who argue that the rate of decrease in T cell maturation in the thymus (exponential with a half life of 15.7 years) can be worked into a model that matches the rate of increase in cancer. As this rate of decrease is different for men and women, you get two different slopes. Not that this model assumes that “intrinsic” cancer risk stays constant through life! That is, that the model doesn’t care about more mutations making it easier for a cell to become cancerous. While the model seems to work, there are a few types of cancer it cannot fit well, which the authors try to justify, caveating that their model tries to capture some aspects of tumorigenesis but not all.. A second model presented there (Power-Law-Immunologic Model, PLIM), that accounts both for a power-law increase in risk due to mutations accumulation and an exponential decrease in immunosurveillance. Unsurprisingly this model works better than any of the other two separately. Looking at the exponents that result from fitting, they conclude that the immunosenescence factor is more important to explain the increase in cancer risk over time. Moreover, the authors compare the PLIM predicts that colon cancer would have a lower number of mutations needed on average for cancer (2.2) while the pure power law model predicts 6.3. As it happens, the empirically measured value is more around 2.3 (per Palmer´s citation) or 3 (per Anandakrishnan et al., 2019).
A second model presented there (Power-Law-Immunologic Model, PLIM), that accounts both for a power-law increase in risk due to mutations accumulation and an exponential decrease in immunosurveillance. Unsurprisingly this model works better than any of the other two separately. Looking at the exponents that result from fitting, they conclude that the immunosenescence factor is more important to explain the increase in cancer risk over time. Moreover, the authors compare the PLIM predicts that colon cancer would have a lower number of mutations needed on average for cancer (2.2) while the pure power law model predicts 6.3. As it happens, the empirically measured value is more around 2.3 (per Palmer´s citation) or 3 (per Anandakrishnan et al., 2019).
The authors are also able to derive from the model a threshold over which the immune system cannot maintain control of cancer, 1e6 cells and provide evidence that in thymectomized mice, their cancer risk increases, while thymus grafts to mice born without a thymus (nude mice) reduces their cancer risk. Anecdotal, in a rare paper that tried two different concentrations on cancer cells injected to mice to induce cancer ~1e5 failed while ~1e6 succeeded (Schmidt and Good 1975), though these were human cancer cells so presumably the immune system of mice had an easier job targeting them.
Not everyone is convinced, Jimenez-Alonso et al. (2018) argue that humans with DiGeorge or complete DiGeorge (complete lack of thymus at birth) syndrome, cancer rates in children are not as high as observed in the elderly despite being more susceptible to infectious disease. Some cancers do not uniformly increase in incidence, contra what the model would predict: testicular cances rises in indicende until 30 years of age, decreasing thereafter, in fact it is extremely rare among the elderly. They also argue that in athymic mice, there is actually no increase in cancer risk if one considers spontaneous cancer, not cancer induced by carcinogens.
Schooling and Zhao (2018), also replying to Palmer et al. suggest that he has overlooked the role of androgens in driving the gender differences he found but don’t directly challenge the key results.
Mortazavi (2018) raises some issues, one is that cancer rates decline or plateau near the end of life, that some cancers are more common in children, that some cancers peak in mid-age (as previously discussed, testicular cancer), or that individuals living in unusually radioactive regions of the planet (like Ramsar in Iran) don’t really get more cancer than the rest of people.
Palmer et al. replied in the same issue of PNAS. They agree that mice evidence is mixed, with thymectomy studies supporting them and congenitally athymic nude mice but say evidence for DiGeorge syndrome is mixed. For the mice they dispute the use of athymic mice to evaluate the model because their lifespan is short, giving less time for cancer to occur. For DiGeorge in humans they say that the extreme case of complete lack of thymus is very rare, and that most DiGeorge cases seem to retain some degree of immunocompetency. They also address the other issues in a way I deem reasonable. However adjudicating who is right about the mice studies would require a more extensive review.
There is also more recent evidence on this, Almeida-Santos et al. (2020) explored, among other things, the effects of thymectomy on cancer progression in young mice, but here the cancer (1e5 cells) was grafted onto them. This number of cells is closer to the threshold identified by Palmer and in any case it is not a situation that an animal would encounter in the wild (1e5 cells suddenly all turning cancerous), so this is evidence that immunosurveillance doesn’t make much of a difference once the cancer is past a certain critical size. They also found that the aged thymus may actually be a net negative for cancer: Thymectomized mice actually responded better to immunotherapy.
Palmer et al. cited Anderson et al. (1978) who did a 2x2 study with (0, 600 rad) irradiation X (thymectomy, sham surgery) study. They also made sure the mice were, as far as they could see, free of any infection, which is important because they want to see what absence of thymus does for cancer once infections are controlled away; lack of thymus would lead to more deaths from infection. They had 277 out of 302 mice that survived initial irradiation. The key result is summarised here: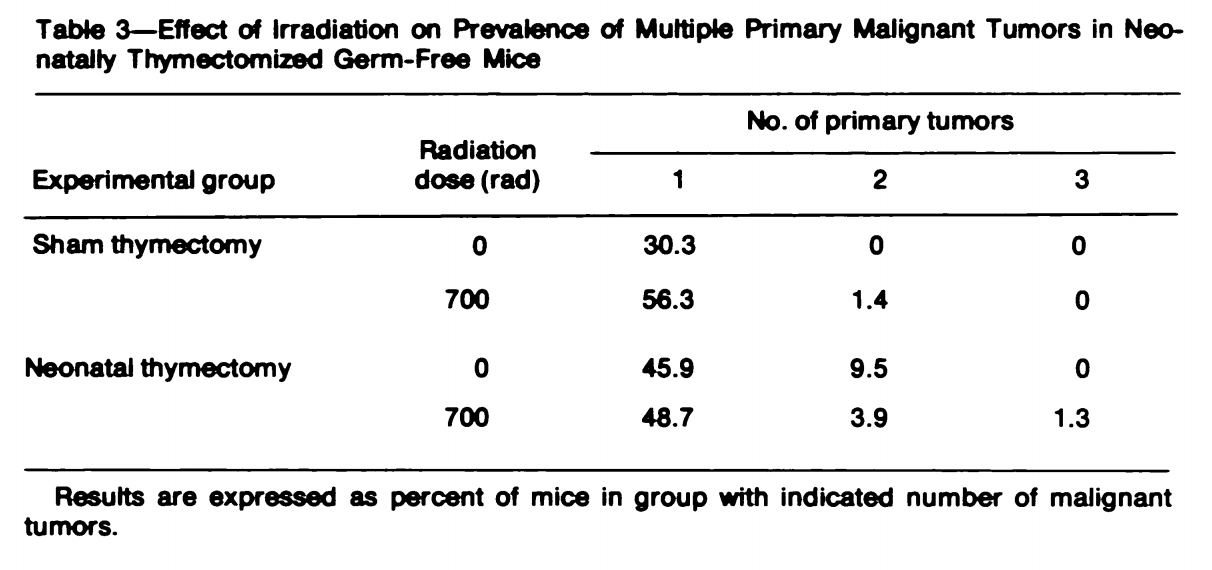 Neonatal thymectomy does increase the percentage of mice that developed a tumor both in the case with and without radiation. Interestingly radiation didn’t seem to matter much for the thymectomized mice, suggesting that the additional mutations did not increase cancer risk above the existing increase from reduced immunosurveillance. But despite developing more cancer, the irradiated mice without thymus had a markedly higher median lifespan than their sham surgery counterparts.
Neonatal thymectomy does increase the percentage of mice that developed a tumor both in the case with and without radiation. Interestingly radiation didn’t seem to matter much for the thymectomized mice, suggesting that the additional mutations did not increase cancer risk above the existing increase from reduced immunosurveillance. But despite developing more cancer, the irradiated mice without thymus had a markedly higher median lifespan than their sham surgery counterparts.
They also cited Miller et al. (1963), where they painted a small (<25) number of mice in a carcinogen and tracked skin cancer occurrence over time. 100 days after the treatment, 50% of the thymectomized mice showed cancer, while only 5% of the regular mice did so. This paper was approvingly cited by Miller (as it happens, the discoverer of the function of the thymus) recently.
And Grant and Miller (1965) who did a similar thing noted that during the first weeks after intramuscular administration of a carcinogen, the thymectomized mice show substantially more cancer, but by the 22nd week they both had converged to similar rates. This illustrates something important to assess whether or not the thymus matters for cancer: As involution occurs, if the Palmer model is right we should expect the same rate of cancers in thymectomized adult and intact mice. In the initial model in the Palmer paper one should indeed observe more cancer in young mice; however the PLIM model would still require time to pass for cancer to occur. With induced carcinogens is different, with an increase mutation rate then it will be easier to actually induce oncogenesis and there will be a chance to see the differences between an active thymus vs an absent one.
In other words, the ideal test of this hypothesis is leaving mice intact and regenerating or transplanting thymuses of adult mice, and comparing then cancer rates.
I could find some review work doing a systematic review of the thymus-cancer connection. The earlier ones here are framed back in the days where the idea of immunosurveillance was not accepted: it used to be believed the immune system had little to do with cancer, so the prior was that the thymus would only affect tumorigenesis via, if any, virus that cause cancer.
Allison & Taylor (1967) while noting heterogeneity in the results, ultimately note that The simplest expectation, that neonatally thymectomized animals would show an increased incidence of many different types of tumors because the tumors are frequently antigenic and thymectomy depresses cell-mediated immunity, is no longer tenable.
Kersey et a. (1973) argue the case is not closed yet, there being sufficient evidence on the immunosurveillance side, and that further work is needed.
By 1976, Stutman in a very lengthy review still says it is not clear, for tumors of viric origin there is sufficient evidence, but for the other cases it’s unclear, further urging researchers to design better experiments, in particular studying spontaneous tumors rather than inducing or grafting them.
Fastforwarding to the present the current consensus seems to be that the nude mice studies were not ideal because they do still have an active immune system, and their usual control group, CBA/H mice, are more vulnerable to chemically induced cancers than the nude mice (because of an overactive enzyme in that particular strain that turns the chemical into its carcinogenic form), and that for the spontaneous tumor development case, the followup periods were not enough (Smyth et al. 2006).
One of the most recent reviews that looks at the role of the thymus in cancer, autoimmune disease and more is a doctoral thesis in German that I didn't fully read (Suchanka, 2015), the author ultimately concludes that Due to a great heterogeneity in study design and study outcome, the findings of this review were reported qualitatively. [...] The findings of this review support the hypothesis that thymectomy may induce premature immune aging with increased risk of autoimmunity and infectious diseases but no increased risk of cancer. Nevertheless, more basic research and longitudinal clinical investigations are needed to prove this hypothesis.
The kind of heterogeneity the authors talks about refers to studies with small samples, poor controls (e.g. surgery vs no sham surgery), poorly controlled pathogenic conditions (which lead to high death rates, biasing the study), and the study doesn’t look at adding back a thymus rather than taking it out.
There is other evidence we can look at is human children that have undergone (partial or complete) thymectomy due to heart surgery. Gudmunsdottir et al. (2018) did that, excluding from their sample DiGeorge individuals as well as those that had received a heart transplant (Which usually involves removing the thymus). As controls they used individuals that had undergone cardiac surgery without thymectomy, or the general population. The author express their reservations about their conclusions; relative to the general population they do find an increase in autoimmune disease and cancer. They followed the subjects for up to 37 years, with a mean of 13 years, so they wouldn’t have time to observe what happens in the old age.
Still this is not ideal because in a true model cancer incidence is due to mutation accumulation (so you need time) and lack of immunosurveillance, which is absent both in the thymectomized individuals as well as the general population when they are adults. What we’d want to see is what happens when the thymus is rejuvenated or prevented from involuting, do we see effects then?
The latest review I could find of the role of the thymus in tumorigenesis is Wang et al. (2020) and largely agrees with the idea that the thymus matters to prevent tumorigenesis, specifically via vastly reduced naive T cell production, which leads to higher % of less effective senescent or exhausted T cells. Senescent or exhausted T cells cannot undergo clonal expansion so even if they could attack cancer, they wouldn’t be able to mount a strong response. To make it worse, with thymic involution, as discussed earlier, the thymus is biased towards the production of Tregs, which put a break to Teff action, further decreasing the effectiveness of the immune system to fight cancer.
Separately there’s evidence from immunosuppressed adults, but this is less clear because we would be comparing not the immune system at its best vs the post-immunosuppression, but with somewhat already impaired adult immune systems. A meta-analysis shows no effects (Shelton et al., 2016) but some complaints that the meta-analysis is biased due to cancer severity affecting if a patient is recommended immunosuppresion (Beaugerie & Peyrin-Biroulet, 2016).
While the findings from Palmer are intriguing, I think the empirical evidence to back the model is not there. At best it's unclear and at worst it falsifies the model. My prediction would then be that if you just rejuvenate the thymus you may get some benefits regarding immunosurveillance but not make cancer rates in the old the same as in the young, as the model would suggest.
Reversing immunosenescence
What could be done to make the immune system young again?
Thymic rejuvenation
FOXN1
Of course, making the thymus great again is one idea, and here perhaps sustaining FOXN1 activity is able to prevent the involution. The answer is somewhat, but not completely. There are reviews of this area in general: Chaudrhy et al. (2017), Lepletier et al., 2015 (from where I took the image below)
Cell therapy
Oh et al. (2020) grafted induced/reprogrammed TECs into old mice (18m), leading to levels of inflammation (As measured by IL-6 and IL-1beta) similar to those of young mice, which represents a large effect (2-3x reduction, Figure 6 in the paper), reducing T cell senescence, and self-reactive T-cells (though not to levels of young animals). This was accompanied by a 2x increase in thymopoiesis which is a large effect. The effects were observed 2 weeks after engraftment
Gene editing
Bredenkamp et al. (2014) using a mouse model that allows transient expression of FOXN1 at will and show that the involution can be reverted by a year (in mice) as measured by thymocyte count or naive T cell %. However, this is way from fully rejuvenating it. If FOXN1 is the culprit then perhaps giving thymic cells more FOXN1 will help.
The only two cases I have been able to find where the thymus does not involute are in the Buffalo/mna rats that present a mutation that leads to an enlarged thymus that does not involute. This is not due to cancerous growth, but merely hyperplasia, and the resulting lymphocyte populations looked normal. Unfortunately the thymus becomes so large that it kills the rats from asphyxia or difficulties eating, but this can be prevented by surgical removal of the pituitary gland (Hirokawa et al., 1990) or thymectomy coupled with implantation of thymic tissue in the kidney capsule. The equivalent gene in humans that is mutated is ACTR3.
The other example is presented in Garfin et al. (2013) where Rb1 knockout leads to thymic hyperplasia too, with the same effects as in the previous study (asphyxia), to the point where 7 month old mice have more thymocytes than 3 week old mice. This was mediated through, among others, an increase of FOXN1.
Thymus transplantation
I mentioned already a few thymus transplantation studies in mice. In humans it is a common procedure for complete DiGeorge syndrome patients, although one not without risks (Markert et al., 2016), it can lead to graft rejection or autoimmune disease. Naturally, the right way to do this would be to use the patient’s own cells to generate a thymus and transplant that.
Bortolomai et al. (2019) seeded a collagen matrix with wild-type adult TECs modified to transiently express Oct4 (the O in OSKM) to promote their proliferation, then implanted it to athymic mice, but despite being able to induce vascularization of the tissue, they did not manage to get it to train T cells, macrophages quickly dismantled the graft. This is not all bad news: After all the artificial thymus they generated had a 3D structure resembling that of the real one. Perhaps with a different matrix (instead of collagen) or more TECs or iTECs from the host animal better results could be achieved.
Epigenetic therapy
If epigenetic alterations (of tissue-resident macrophages or HSCs) are a root cause, targeted epigenetic reprogramming to rejuvenate them would be a therapy but I couldn’t find any studies that looked at this in particular. There are studies that do whole-animal epigenetic rejuvenation as in the Izpisua-Belmonte lab work from 2017 and the only immune system-relevant results they report is a recovery of the spleen, to levels intermediate between old and young specimens. There is no other experimental work I could find that looked at immune-system effects of epigenetic reprogramming.
Various cytokines
IL-21 administration showed promising effects in old (15m) but not young (2m) mice, as measured by T-cell population composition, total thymocyte count, naive T cell %, and TCR diversity a week after (Al-Chami et al. 2016). Given that IL-21 promotes T cell proliferation it is important to see if these gains are actually from the thymus or not just proliferation of the already circulating T cells. It has to be the thymus: TCR diversity cannot be increased by naive cell clonal expansion, effector T cell % went down, and specifically recent thymic emigrant % went up, so regardless of proliferation, the thymus also increased its output.In a less natural context, IL-21 is also able to produce thymic regeneration after damaging it with glucocorticoids (Rafei et al., 2013)
IL-7 is another cytokine that has likewise shown some promise (Chaudhry et al, 2017) but in humans, at least in a bone marrow transplantation context (Perales et al., 2012) the effects seem to mostly cause an expansion of already present T cells, not a substantial increase in generation of new naive ones in the thymus.
Heterochronic parabiosis
The bloodboys follow us around everywhere we go, we are always asked about the bloodboys” - Laura Deming
Whether replacing old with young blood in old mice helps with the immune system is something that naturally comes to mind when talking about the immune system in a longevity context. Obviously if you infuse young mice T cells that is going to have some effect. But does it do anything to the thymus? The answer seems to be no, albeit only a few studies seems to have looked into this ( Kim et al. (2015), Davies et al. (2015), Pishel et al., (2012)).
These studies however can also be used to see if T cells from old individuals can work if exposed to a non-involuted thymus, and here the answer is yes, they can.
The role of zinc & other micronutrients
As we age, circulating zinc in blood decreases (and copper increases) and the ratio between copper and zinc has been associated with various kinds of disease and which has been attributed to various causes (Malavolta et al. 2015, Romero Cabrera (2015)).
- Inflammation
- Decreased expression of the ZIP proteins, which transport zinc into the cells, both in enterocytes in the intestine but also in lymphocytes themselves; or increased expression of ZnT which do the opposite, the cause of which seems to be epigenetic in nature (Wong et al., 2014)
- Hormonal changes, in particular the loss of nocturnal spikes in growth hormone and in turn IGF1 or as part of an programmed regulatory response, as Cu has antimicrobial properties which fits with the idea that the body favors the innate immune system at the expense of the adaptive with aging.
Mocchegiani et al. (1994) study the effects of zinc supplementation in mice. Zinc is necessary for the activity of thymulin, a peptide secreted by the thymus. With age, the levels of zinc in blood, both in humans and mice decrease due to a lower expression of zinc transporters in the cellular membrane. In the elderly, the levels of circulating thymulin are similar to those in young people, but the active fraction (bound to Zinc) does decrease.In young mice, circulating zinc levels are around 114 ug/dl, with 81 for old mice and 91 for the treated mice. The fraction of zinc-bound thymulin in old (24m) treated mice was found to be equal to those of adult (6m) mice. The result of this was a regrowth of the thymus to a size 75% that of young mice which is almost twice as large as that of old untreated mice. Thymocyte counts increased by a factor of 6 relative to untreated mice, ultimately becoming 26% lower than in younger mice. NK cells cultivated in vitro were twice as able to eliminate lymphoma cells (but still half as able as those of young mice).
That is not the only study looking at this: Wong et al. (2009) gave mice 300 mg/kg zinc to old mice resulting in 52% more thymocytes; a valus still 2.5x lower than in younger individuals. These effects are smaller than in the Mocchegiani study. This doesn’t seem to be because of a lower dose of zinc: plasma levels of zinc were similar; rather it to be because Wong used very young mice (8 weeks) while Mocchegiani used adults.
Prasad et al. (2007) supplemented a sample of elderly individuals with 45 mg of zinc per day and found a substantial reduction in infections or symptoms like fever over a year.The Cochrane Collaboration (2013) has a systematic review of the use of zinc (>75mg/day) for the common cold, finding that it reduces the duration of common cold symptoms as well as reduces their incidence but not the symptoms. The authors think more evidence is needed, so they wouldn’t support recommending zinc as a prophylactic.The effect is greater in adults than in children, but the review didn’t look at the elderly, who probably would derive the greatest benefit.
I single out zinc because it seems like it shows the most robust effects but it’s not the only micronutrient that has been tried, the same seems to be true for high doses of Vitamin C (Ran et al., 2018), who recommend a daily dose of up to 1g and a larger (3/4g) dose during infection with the common cold. The latest Cochrane review (2016) reached similar conclusions, if perhaps being more cautious. Neither of these reviews looks at the elderly in particular.
Combined therapy
If you have read my longevity FAQ you probably know I like combined therapies.
In humans, Greg Fahy’s 2019 Reversal of epigenetic aging and immunosenescent trends in humans is probably the most successful application of combined therapies to treat immunosenescence in healthy volunteers. The sample size was small and the treatment was adjusted on an individual basis and no one has done anything similar. The same team will be doing a larger trial (TRIIM-X) soon to validate the results.
The treatment here was a combination of human growth hormone, DHEA, metformin, (these, ¾ times a week), zinc (50mg daily) and Vitamin D3 (3000 IU, daily). DHEA and metformin are there to counteract the increase in glucose and insulin levels that GH induces.
They found a decrease in CRP, but not IL-6 (Which did rise at the beginning of the treatment but went down afterwards). A few side effects common of GH administration were reported in some cases (joint pain, anxiety, or carpal tunnel syndrome). Glucose levels and insulin did go up a bit, but stayed within normal parameters.
The key improvements were:
- The mass of the thymus, in one participant the fat free fraction almost doubled in size, although this was in a participant with a highly involuted thymus in comparison to others.
- Monocyte % went down by half, with the lymphocyte to monocyte ratio subsequently increasing by half too. Presumably this was because of an increase in lymphocytes. This stayed constant 6 months after the trial.
- The fraction of monocytes expressing CD38 (perhaps a proxy for M1 macrophages) went down by 40%, and so it is likely that this improved NAD levels too.
- The % of naive CD8 and CD4 cells increased; but it went back to normal at the end of the trial
- PD1+ CD8 (exhausted) T cells went down by 20%
- No changes in circulating senescent T cells
- And the highlight of the study, lifespan increased by 2 years according to the GrimAge clock, and this stayed the same after discontinuing treatment
Note that they measured senescent cells as expressing CD57 or not expressing CD28 but (Martinez-Zamudio et al., 2020) warn of the dangers of this sort of things:A primary reason for this uncertainty is that specific markers used to identify senescent CD8+ T cells in the past (19, 20), such as a loss of the cell surface receptors CD28 and CD27 and a gain of expression of CD45RA, CD57, TIGIT and/or KLRG1 do not accurately characterize all T cells that have permanently lost the ability to proliferate due to acquisition of macromolecular damage, upregulation of cyclin-dependent kinase (CDK) inhibitors, and development of senescence associated ß-Galactosidase (SA-ßGal), criteria that define the state of cellular senescence (16). In fact, CD8+ T cells that have lost expression of CD28 and/or that display varying levels of CD45RA, CD57, TIGIT or KLRG1 maintain the ability to proliferate following appropriate stimulation, which is incompatible with a classical senescence response (18, 19).
This really sounds promising. If one is willing to trust GrimAge and some of the previous mice studies then it does look like a robust result.There is one caveat one could make: It could be argued that the study just rejuvenated the immune system and nothing else. The GrimAge clock was applied to the blood, not the liver or the brain so it may well be that those tissues are as old as they started; in turn this could reduce the predictive power of GrimAge: It is likely that in natural aging there is a structure of correlations between blood-based methylation and methylation in other organs, with each aging at its own pace, but with a novel procedure like this, this structure could be altered so that rejuvenated blood no longer implies everything else is rejuvenated too; in turn this would void GrimAge’s predictive power. To be sure, the changes observed were in the right direction: The reduction in CD38 monocytes and increase in LMR ratio are already good signs; and if we accept that the immune system is deeply connected to other hallmarks of aging, then it would make sense to infer that actually this went beyond merely the measured biomarkers.
Others
Alterations in the extracellular matrix have been recently proposed as a hallmark of aging. As impaired intercellular communication seems core to immunosenescence, it may be that rejuvenating that also helps.
Conclusion
Immunosenescence is a complex phenomenon! The immune system is entangled with the rest of the aging process in a way that makes it hard to neatly untangle. However, there are some things that can be glanced from the dozens of papers that I’ve read to write this.
First is that there seem to be some root causes: Some are damage related (In HSCs and potentially tissue-resident macrophages), others are programmed (involution of various tissues). It may well be that addressing the programmed causes also fixes the damage ones, depending on how much do intrinsic vs extrinsic factors matter for immunosenescence, a debate that remains unsettled.
Second, is the importance of intercellular signaling: Cells may be okay but act old if surrounded by old cells. Therapeutically, this suggests that you would need to replace the entire microenvironment of the cell for the greatest impact. In the same way that the most rapid way to change culture is rapidly replace the people that embody that culture, the fastest way to get old cells to act young is to rejuvenate a large amount of them, simultaneously. Rejuvenating just the thymus or just engrafting some young HSCs will not be enough.
Third, is that knowledge in this area seems to be changing too. Back a few years ago the view was more skewed towards cell-intrinsic damage, a central role of the involution of the thymus, and large drops in naive T cells and TCR diversity, with the CMV as a key driver. Relative to then, it seems all those things are less relevant, with the signaling story getting more support.
Fourth is the incredible diversity there is in the immune system, of an almost quasi-fractal nature. Every year new subpopulations are being discovered and what seemed to be dogma is then challenged and replaced. Many reviews stated that we are just starting to know more about immunosenescence in particular and that forming a clear picture has been made difficult by conflicting findings due to all these highly specific subpopulations. This diversity extends across species: mice immune system dynamics are not like those of humans.
Fifth is that, in my view, a factor that has severely slowed down progress here is a combination of lack of standardization and reluctance to thoroughly answer concrete questions, leaving them at “there is conflicting evidence”. Ideally, for concrete questions like “Do HSC numbers increase over time?” or “Does the thymus play a large role in cancer surveillance?” we would have some central repository -or at least a meta-analysis or meta-regression study- that tabulates all the factors that cause studies to disagree with each other, and spot if it is just less precise assays that are the cause of the discrepant findings or if there are genuine differences under the noise. Lack of standardization affects the field as a whole because it makes hard to draw conclusions from a pool of findings, having to resort on extrapolations out of conflicting results. Standardization would include an agreed battery of general immunocompetency, to be assayed in the same way in every study. No more “in-house markers our lab likes to see if something is a senescent cell or if a T cell is naive or not”.
Sixth is that given that interactions between the components of the immune system are incredibly complex, but arise out of a substantially smaller set of causes (involution of the thymus, spleen, adrenals, maybe HSC and macrophage epigenetic noise, etc), rather than continuing to document all the ways in which the components loss their function, the need for root-cause analysis and combined interventions deserves more attention. It is fine if the degradation of function has no free-standing root cause and instead is a convoluted mesh of feedback loops, with no key players, but still identifying them and being able to treat all of them at once in vivo should lead to faster progress in reverting immunosenescence.
Seventh, understanding and ultimately reversing immunosenescence in isolation, as this review I hope has shown, is hopeless. For example, to fully explore the hypothesis that the behaviour of dendritic cells are a larger factor than commonly thought one would need to look at the extracellular matrix e secondary lymphoid organs-and how to rejuvenate that. Or to study the role of reduced zinc on the immune system one has to look at why is there reduced absorption in the first place. Or to see if leaks in the gut are a large contributor to inflammation, one needs to study why that happens and how to revert it. Evolutionary dynamics cannot be disregarded either. In the same way cancer evolves to outcompete the immune system, it can also be -as noted in the stem cell clonal diversity case- that populations within the immune system exist in a state of cold war with each other, vying for spreading their numbers at the expense of others.
And eighth, the combination of therapies that would seem ideal to test would be (assuming away safety concerns): thymic rejuvenation (Fahy cocktail, iTECs infused with FOXN1 etc) to get fresh naive cells circulating, whole-body (or if possible, targeted to stem cells) epigenetic reprogramming, crosslink breakers to rejuvenate lymph nodes and facilitate T cell motility and DC activity, adrenal rejuvenation to get DHEA back perhaps.
Appendix
There are also studies that looked at the effects of gender or things like castration (it causes the thymus to regrow a bit, but then it involutes); I have ignored those as the phenomenon occurs in all members of all vertebrates, and I was interested in the shared mechanisms; for therapeutic use it may make sense to take this information into account when designing a treatment but that is not the object of this article. Another topic of interest that I didn’t cover is the aging of the spleen and lymph nodes. It is an under-researched topic that should get more attention, but the article was becoming too long already so I cut that out.
Here are some other studies I looked at that informed the article but that didn’t add to what was already cited, or that contained information outside of the scope of the review.
- Transplantation of newborn thymus plus hematopoietic stem cells can rescue supralethally irradiated mice
- Known unknowns: How might the persistent herpesvirome shape immunity and aging?
- Interleukin-21 promotes thymopoiesis recovery following hematopoietic stem cell transplantation
- Altered Thymic Activity in Early Life: How Does It Affect the Immune System in Young Adults?
- Stem Cells in Diseases of Aging
- Altered glucocorticoid metabolism represents a feature of macroph‐aging
- The Ins and Outs of Thymic Epithelial Cell Differentiation and Function
- Transgenic Exosomes for Thymus Regeneration | Immunology
- Thymus Regeneration and Future Challenges
- Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention
- Human Inflammaging
- Lymph nodes as barriers to T‐cell rejuvenation in aging mice and nonhuman primates
- Functional and Homeostatic Impact of Age-Related Changes in Lymph Node Stroma
- Immune senescence: significance of the stromal microenvironment
- Cancer and Aging - the Inflammatory Connection
- Image-guided intrathymic injection of multipotent stem cells supports lifelong T-cell immunity and facilitates targeted immunotherapy | Blood | American Society of Hematology
- Thymus Size and Age-related Thymic Involution: Early Programming, Sexual Dimorphism, Progenitors and Stroma
- Impact of aging immune system on neurodegeneration and potential immunotherapies
- Modulation of dendritic cell and T cell cross-talk during aging: The potential role of checkpoint inhibitory molecules
- Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging
In addition to that, the initial set of papers that seeded this post can be found here
Changelog
- 2020-07-28: Added section on Tregs
Citation
In academic work, please cite this essay as:
Ricón, José Luis, “Immunosenescence: a review”, Nintil (2020-07-25), available at https://nintil.com/immunosenescence/.
Backlinks
- Aging is already solved in vitro. What comes next?
- How to solve aging
- Images and Words: AI in 2026
- Beyond lifespan as a metric in aging research: why reprogramming is promising
- Links (40)
- Links (52)
- Links (53)
- The Longevity FAQ
- Tools for knowledge
- What is aging?
- What should you remember? SRSing your life
Comments