From chaos, order: On the nature and measurement of biological aging
Table of Contents
- Aging by the numbers
- Here, have a map
- Causality in biology
- Environmental vs intrinsic aging
- Clocks and their weights: DunedinPACE
- Doing (aging prediction) by non-doing (aging prediction)
- Everything is connected: epigenetic clocks are not unique, but they are convenient
- Epigenetic clocks and organismal aging
- Do neurons age?
- Better is worse: desiderata for a gold standard aging predictor
- Somatic mutation accumulation correlates with aging
- Axolotls!
- Beyond the epigenome: cancer and telomeres
- iPSC reprogramming as a test for aging measurements
- Aging: stochastic vs deterministic
- Car aging as a way to understand human aging
- Methylation turns over: cars at not cells
- Aging as entropy
- Stochastic aging clocks
- The epigenetic magic wand: a thought experiment
- The intrinsic randomness in lifespan
- Aging interventions that slow down cellular aging are mostly DNA damage reducing interventions
- Appendix: Proteomic clocks
- Summary
What is aging and how to measure it is an everpresent question in the field of aging research. Given the complexity of biology many give up on the task, proclaiming that "we" (either the field or humanity) don't understand aging. I don't. To me, what aging is is clear enough, and we can understand it as a fractal and emergent phenomenon within a system: there's aging of DNA, aging of cells, aging of organs, aging of organisms.
To understand aging and measure it we have to be reasonably acquainted with the systems we are studying, to come up with mental models and validate them against published evidence. The understanding required does not have to be complete and I of course do not claim to understand all of biology, but after having spent a long time thinking about the topic, what follows below is what seems most coherent with that evidence. In classic Nintil fashion, I aim to reach as close as possible conclusions that summarize and harmonize all previously published work by anyone who has ever had thoughts on the matter. This doesn't mean I have read everything, at some point one has to declare it sufficient and assume that what has not been read will still cohere with the model here presented.
I have in the past written some blogposts about aging that are useful to read before this one:
- Epigenetic clocks: A review
- Telomeres: everything you always wanted to know
- Aging is already solved in vitro. What comes next?
- Beyond lifespan as a metric in aging research: why reprogramming is promising.
- Making Cells Young
- What is aging?
The present post is an update of my views on the topic and any discrepancy between this and my previous posts should be resolved in favor of the current one.
Below I present different lines of evidence under the different sections. I tried to linearize it somewhat but ultimately the nature of knowledge is that of a web, not of a linear flow so I haven't tried to coerce too much it into what it is not. I recommend reading this to the end and taking it as a whole instead of being fixated by any one specific section. Some sections may seen like they simplify a lot, but if I had to stop to fully explain everything, it would lose focus.
Before going into it it is useful for me to say roughly what this is about, and what this is not about:
-
Here I cover extensively methylation clocks results and whether they are a good proxy for aging, but I don't do this because I think they are in any way special; rather I do it because they are thematically relevant. They are used by consumers and in research.
-
I discuss what I mean by 'aging' as it applies to various systems, including inanimate objects like a car or a glass of boiling water
-
I reach slightly beyond methylation into proteomics to speculate about the results that might come into the future and what those might enable.
-
I consider the case of iPSC reprogramming and what that might say about aging and the possibility of its reversal
-
I do not consider in any depth the idea that aging is programmed (ie that we evolved purposefully to age in a group selection way) as I consider that to have been sufficiently argued against by others elsewhere.
- While at the same time agreeing that while aging is a universal entropic phenomenon, its translation into phenotypes (how cells and their programs react to the damage) is encoded in the genome of each species and in this sense, it is programmed.
- That is, the 'aging phenotype' of an individual is a function of chronological time, the environment, and its genome.
-
I do not go in depth into the specifics of human aging (e.g. cancer in humans or cardiovascular disease). I focus mostly on cellular aging.
-
I do not claim to have invented much here de novo, and my thoughts on the subject owe a lot to people like Gladyshev and Hayflick (Aging theory in general), Ocampo (Heterochromatin loss), Gorbunova, Seluanov, and Sinclair (DNA damage and its connection to aging), Kenyon (Genetics of aging), Levin (morphostasis) and Alon (Systems biology), and many others.
-
At the end I have more concrete summary that hopefully is more actionable for specific applications
Aging by the numbers
For recreational reasons, I did an epigenetic aging test.
I sent a small sample of blood to TrueDiagnostic, a company that runs epigenetic clocks for consumers, and got back some results. I got the following results:
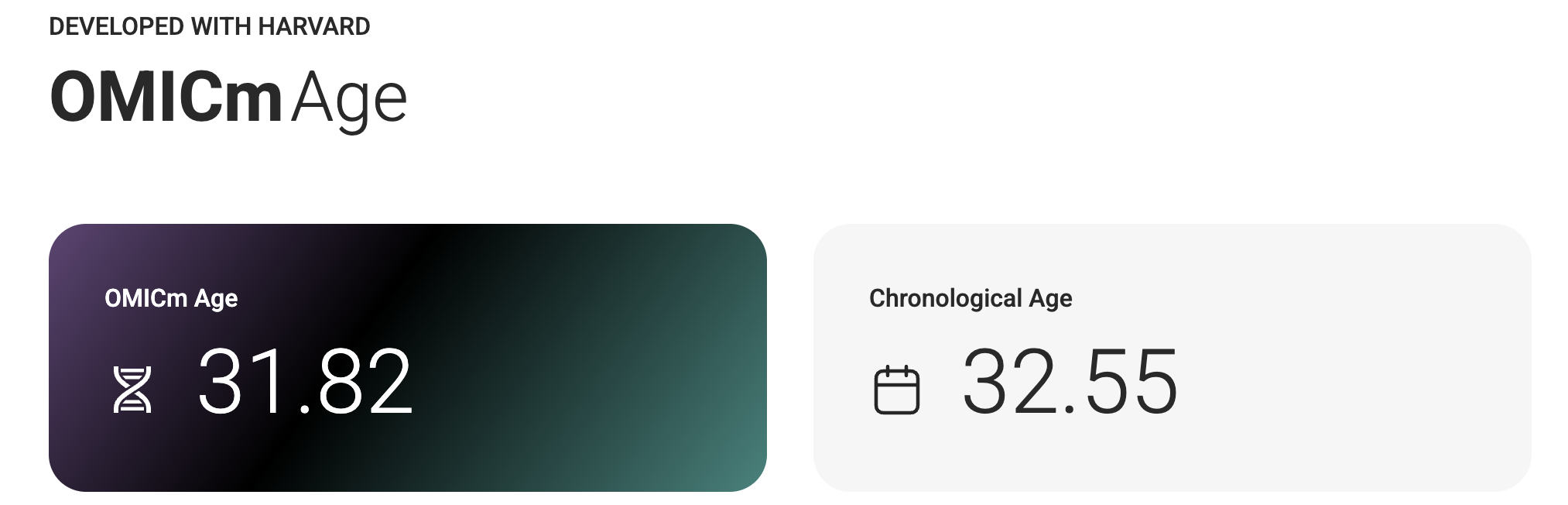
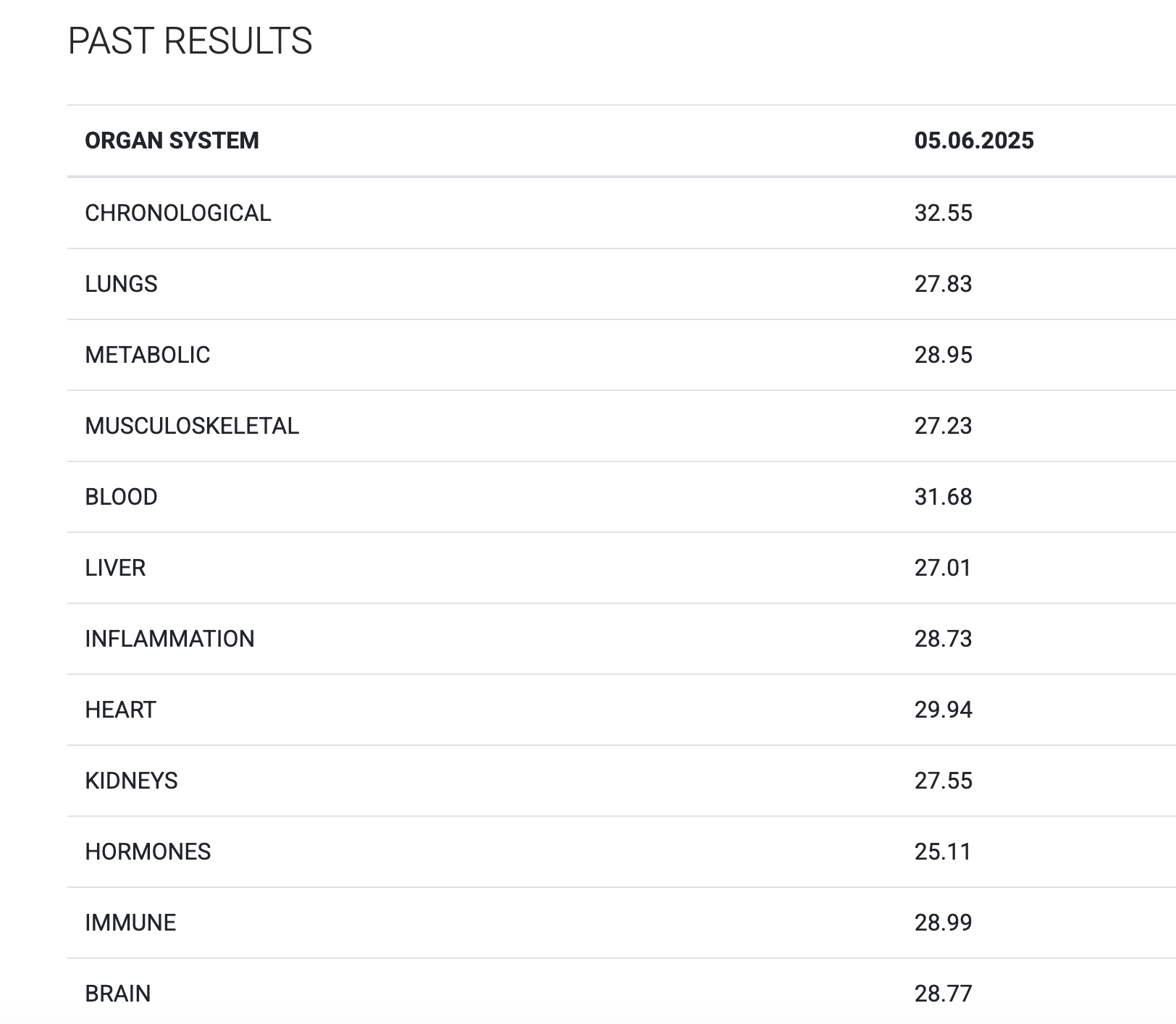
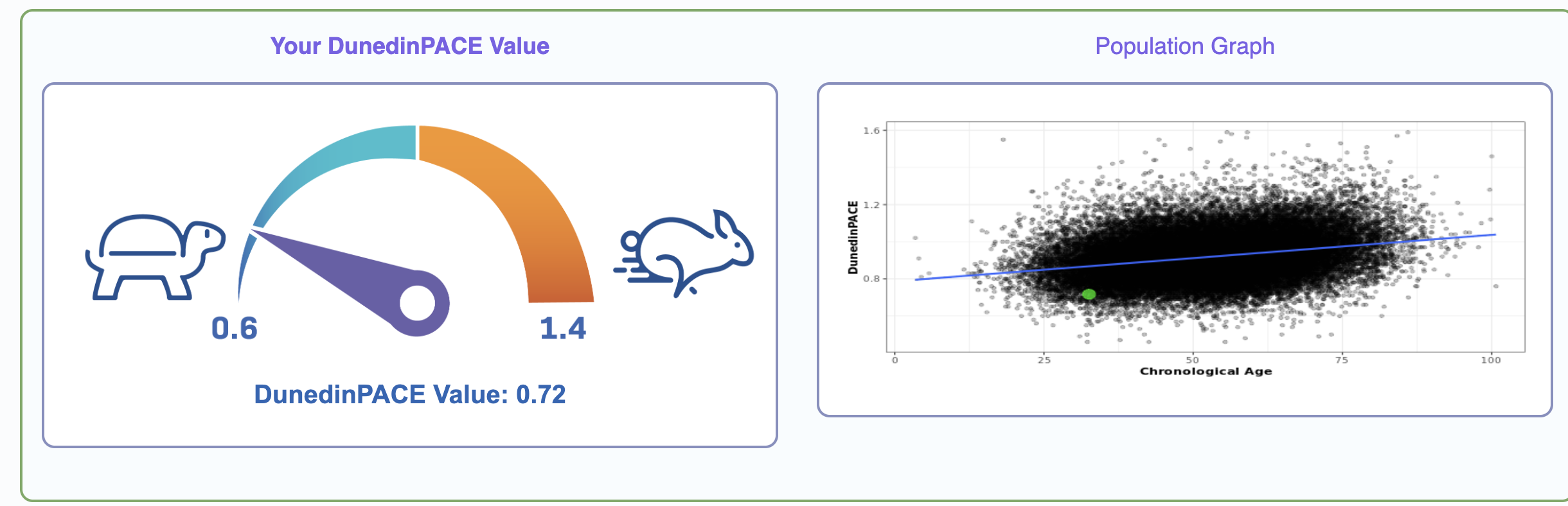
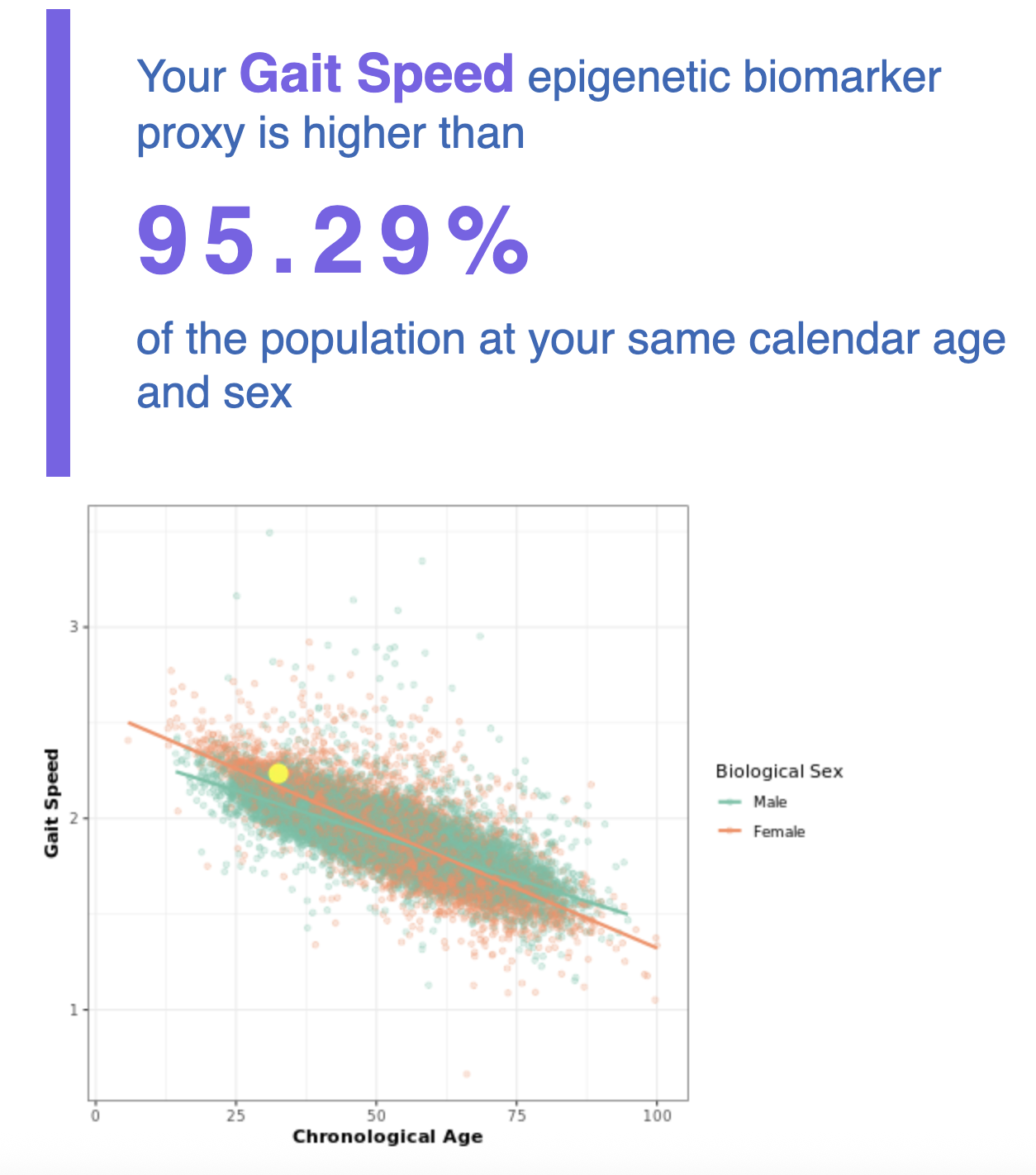
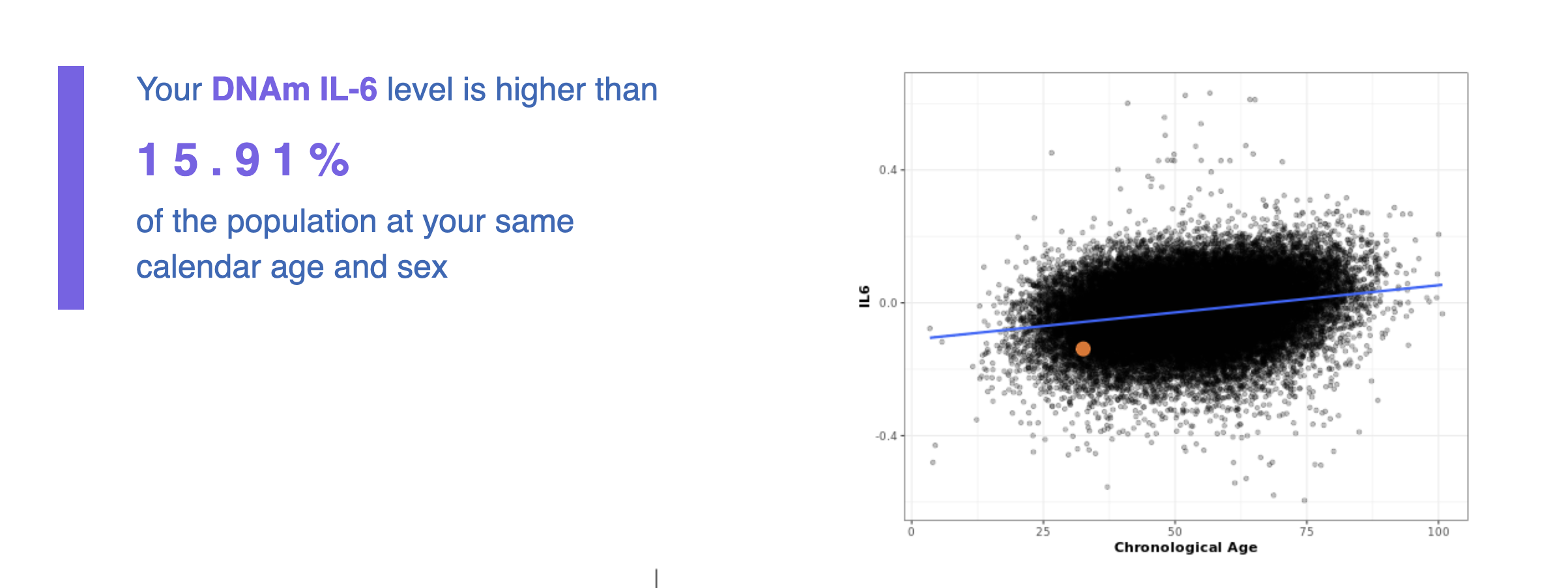
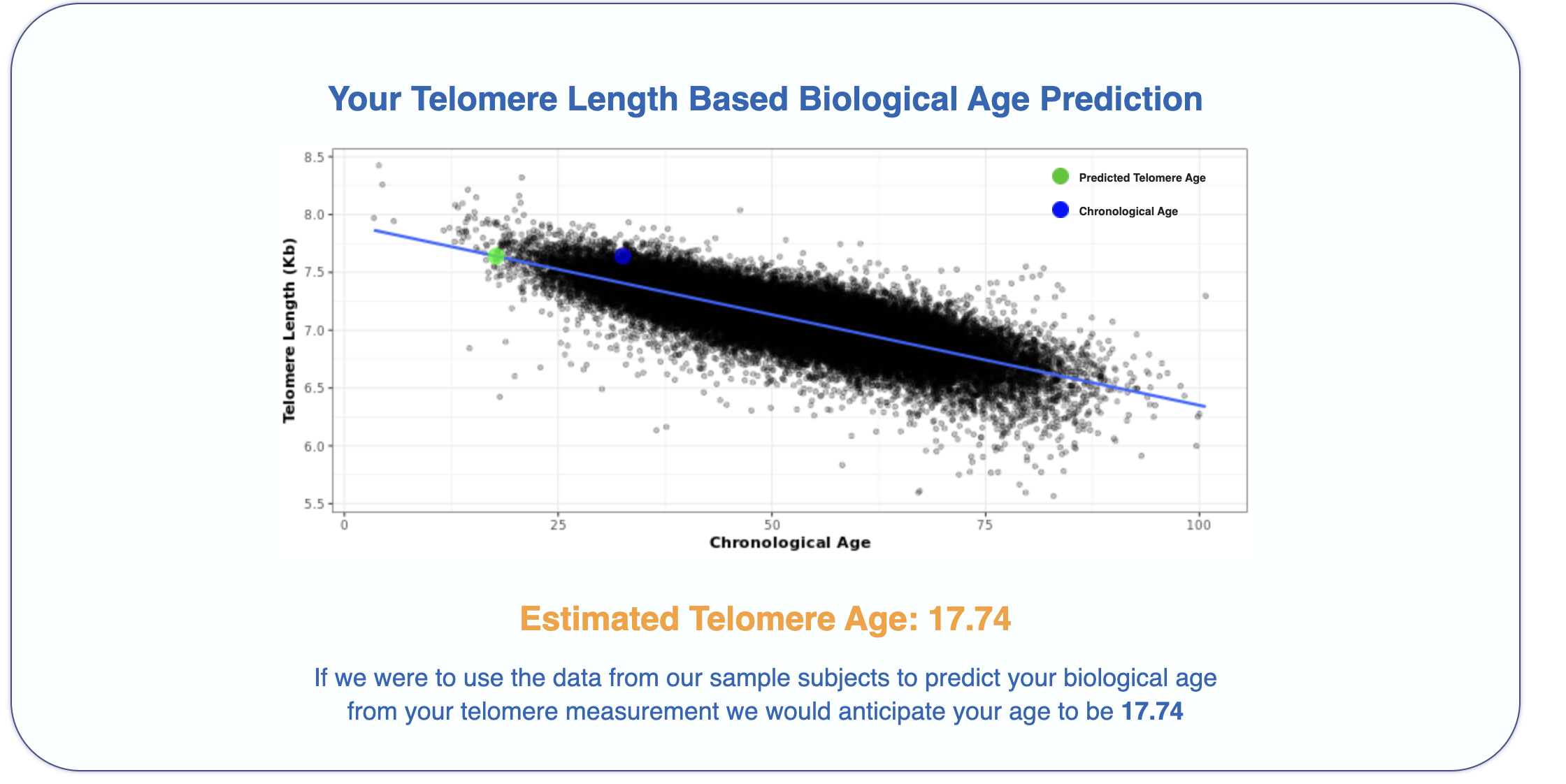
It's interesting that just from a blood sample one can get my actual chronological age within just 1 year, impressive isn't it!
What is going on in the charts above? One says my "telomere age is 17" and another that my "OMICage is 31.82", that my "rate of aging is 0.72" or that my "Gait speed epigenetic biomarker is higher than 95% of people", what does that mean? Is my biological age supposed to be 17? 31.82? Neither? All?
Epigenetic clocks are simple (linear, most often) models trained on the methylation profile of a training set (very often cells collected from blood) to predict some outcome of interest. This outcome could be:
- Chronological age (Perhaps the most common one used, like the OG Horvath or Hannum clocks)
- Mortality risk (As in GrimAge or GrimAge2)
- Some composite endpoint of multifunctional health (Like PhenoAge)
- Here we take a number of parameters like grip strength or VO2max and then we make them into an index (this can all be done independent of chronological age). Then, we can train models that predict grip strength or VO2max from methylation and in turn reconstruct the composite index from a blood sample.
- This is a better approximation to the question "Relative to a reference population, how functional am I overall?" than using chronological age as predicted variable.
- This is not a new idea: Whether someone is aging fast or slow relative to some established biomarker can be easily measured and aggregtead and ways to do this have been around for a while: Klemera-Doubal (2006) as a general way to do this sort of thing and Pace of Aging (2015) are two examples; the latter is what was used to train the DunedinPACE clock. For Pace of Aging it's things like cholesterol, CRP (inflammation), Hb1ac (diabetes), the waist to hip ratio, forced expiration, BMI, etc. But one could do others: There's something called intrinsic capacity from the WHO that sounds to me like a proxy for "aging": it is a framework comprising a set of faculties (locomotion, cognition, vitality (eg muscle strength), sensory, and psychological (wellbeing)). Intrinsic capacity predicts mortality and declines with age as one'd expect. One could also build an epigenetic clock that predicts that too but no one has yet.
- Rate of aging (Like DunedinPoAM or DunedinPACE). This one is particularly interesting and perhaps unique among the clocks beacuse of the way it was built, out of a multi-decade cohort and periodic blood sampling of the same individuals. Here they try to predict not how old you are now but how fast you are getting older; to construct it they also built these composite endpoints as in PhenoAge.
- In theory this means that if I test 10 years from now, my DunedinPACE score might be exactly the same but my biological age (by some clock) will be only 7.2 years older instead of 10
- Their datasets allows this clock to work around an issue the original clocks had: that training on longitudinal datasets could introduce a composition bias, as only the healthiest individuals would be represented at the later timepoints, as the sickest ones would die.
- Molecular markers like telomere length, inflammatory cytokines (IL-6), or cholesterol etc can all be proxied, in theory, via methylation-based models. Per some clockmakers, this is better than using the raw values of the marker because they can be noisier in the short term (IL-6 in particular, I'm looking at you). This is similar to how HbA1c is a more estable measure of diabetes risk than just measuring blood glucose, which tends to vary a lot.
- These markers, to some extent, can be influenced by cell type composition. Someone with a greater proportion of naive CD8 T-cells (that have divided less) will present with longer telomeres if one measures telomeres from blood, so this may be a correlate of "lower lifetime exposure to infectious disease" as opposed to "slower aging", but it could also be a correlate of a more efficient immune system, and that might bona fide be a contributor to slower aging.
- GrimAge2 uses as intermediate markers things like B2M (inflammation-related) or HbA1c (diabetes) as explicit intermediates which skews this clock towards diseases1
as opposed to cellular aging.[1]. What is cellular aging and what is disease? Good question, more on that later
Here, have a map
Before continuing, the diagram below will be helpful for you to understand how I'm thinking about my core model of aging. This is written mostly with cellular aging in mind but it broadly generalizes: at its core sits DNA (nuclear and mitochondrial) from which everything else flows, and the code that helps cells interpret DNA (the chromatin state) which is comprised of modifications of DNA (like methylation) and their supporting structure (histone marks).
These together condition what tools a cell has available (if a gene is broken the cell can't use it) to do its job and how effectively, efficiently, and quickly they can de deployed to react to its environment. Empirically, it seems to be the case that it is chromatin disorganization that by far dominates in the aging process and hence iPSC reprogramming is able to reverse most of it which is quite convenient. Telomere shortening happens naturally when cells divide but it is also a function of our genetics (we could have been born with active telomerase). Through life, the cell constantly reacts to damage, and the response to this damage creates an epigenetic memory that eventually locks the cells in a state of constant inflammation (Patrick et al. 2024), which shifts resources away from the job of the cell (fibroblasts make less collagen, hepatocytes metabolize slower, etc).
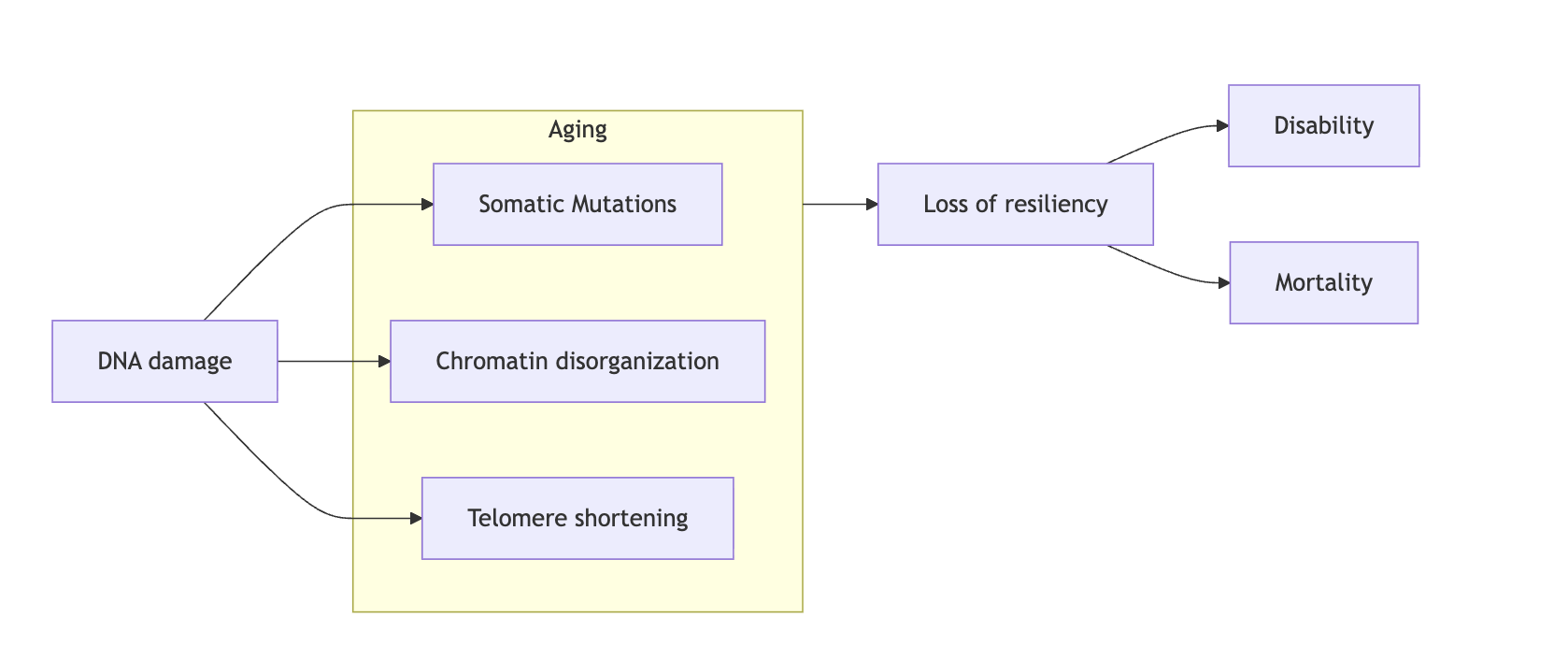
So indeed if cells were able to repair their DNA damage faster than it gets produced, and were able to keep their telomeres from eroding, they would not meaningfully age. They would still get somatic mutations and that may eventually, absent cell division and natural selection of fit enough cells, end up in death. Induced pluripotent stem cells, which don't seem to age epigenetically in culture, do inded drastically upregulate all DNA damage repair pathways (Liedtke et al. 2015). This is existence proof that within our genomes are they are, without any editing, there is enough repair capacity to stave off damage for a long time. One potential wrinkle to this is the case of cells that don't divide; these accumulate age-related misfolded proteins and aggregates that some argue are not digestable even by young cells. Not much research has been done on actually testing it as most research is done on dividing cells. The one paper I am aware of that tested reprogramming in non-dividing cells did see a lowering of one such pigment, lipofuscin (Ivanova et al. 2024). The jury is still out on whether epigenetic rejuvenation can comprehensively reverse this too absent cell division but I am optimistic.
Causality in biology
In biology, everything may potentially cause everything else. Feedback loops abound and disturbing one node enough will eventually disturb the rest. Impair autophagy and you probably get more DNA damage, induce DNA damage and you probably impair autophagy.
As a result, interventions targeting one aspect of the biology of aging can potentially ameliorate impairments in the rest, which can lead one to the wrong conclusion that they are all the same and there are no privileged hallmarks. "Impaired proteostasis" is more caused by "epigenetic alterations" than it is the other way around, for example.
To counter this idea and to defend a more hierarchical model with things resembling root causes, I want to point at the notion of turnover rates: most things in our body turn over. Proteins are produced and degraded, and the same goes for mRNA and most (but not all) cells. Even fibrosis turns over! That's right: fibrosis exists as a balance of generation of collagen and its degradation by macrophages, with a steady state equilibrium of "there's fibrosis" as opposed to the one where "there's no scar" (Alon, 2023).
The epigenome turns over as well: methylation marks are constantly being removed and added back. Methylation gets a lot of airtime because it is easier to measure than what argueably is the most important part of the epigenome: histone marks. These also turn over: they are deposited and removed. This may sound puzzling at first: if the cell is able to just put back marks "where it should" (ie to where young cells have them) why isn't it doing it? And the answer is that they do, but not perfectly, hence a constant ratchet towards aging.
Then there's the genome. The genome gets mutations and chromosomes get messed up to some extent with aging. But those mutations just a tiny fraction of a gigantic set of information. DNA gets copied with relatively high fidelity but it doesn't turn over much.
We should think of the genome not as a code but rather as a vast workshop with miriads of redundant tools the cell uses to do what it does. The epigenome is more aptly thought of as that code: the epigenome is the instruction set that's running on the workshop, determining whether the shop will be making neuron stuff or liver stuff, or aged stuff or young stuff.
There is then a hierarchy of turnover rates:
- DNA: ~almost no turnover informationally, though DNA gets copied (cell division happens every ~20hr)
- mRNA: 10 hr (Yang et al., 2003) to 16.4h (Wang et al. 2022)
- Proteins: From 30 min (Ornithine decarboxylase) to crystallins in the lenses (almost no turnover) as well as histones
- Mitochondria: Days to months ( this and Poovathingal et al. 2021) though these also come with their own DNA (mtDNA) so in a way they are like DNA
- Cells: Neutrophils (6-8 hours) to neurons (for life)
My claim: The slower the turnover rate of something is, the more it matters for aging. Turnover rate is like gravity in a way: Gravity extends infinitely in all directions, everything pulls on everything, but some objects pull more than others. Slow turnover rate of a biological entity is like its mass.
Think of a simple case: If you alter the proteome of a cell a little bit, briefly (you add 1 misfolded protein, other than a prion), what happens next? The cell will go back to equilibrium, degrading the old proteins and making new fresh ones. But if you were to wave that magic wand and set the entire epigenome to an aged state, then for a few hours or days you'd have an aged epigenome and some youthful proteins, but eventually given turnover rates, the aged epigenome would stay and the proteome would then be aged.
The heritability of longevity is higher than what we thought in the past (up to 50% per Shenhar et al. 2025 vs 10-30% I reported back then), and as I discuss in my Longevity FAQ it has been possible to make other species like worms live longer by 10x through manipulating their genome. Given that we already know a lot about the genetics of aging, one could make humans really long lived and slow aging by editing a handful of genes like FOXO3 or CIRBP. Genetic engineering is the easiest way to make something live longer: longevity is encoded in the genome, but for those already born and until we solve bodywide gene editing, rejuvenation of the epigenome is the second best thing we can hope.
Environmental vs intrinsic aging
This fact, that given a normal environment for a species, its lifespan is determined mostly by genetics (compare say a mouse vs a bowhead whale) is phrased by some (like Cynthia Kenyon) as aging being programmed. This is a different use of the term from those that argue that "there's an aging program that purposefully drives the organism towards aging".
The phrase "normal environment for a species" is doing some work there. For many years cardiovascular disease has been a key component of human aging. But suppose that statins, PCSK9 inhibitors and other such interventions manage to make CVD a rare disease. Humans now live longer. Suppose that we also manage to have perfect cancer treatements and we also cure cancer. Now humans live 10 years longer. In that case we would be leaving a central part of what aging is untouched: the thing that human and worm and mouse aging have in common.
That part is cellular aging. Whereas in multicellular organisms new ways of aging arise, for any one single cell, regardless of whether it is yeast or a mammalian cell or a worm cell, they all age in a similar way. Roughly speaking, the main thing that goes on in cellular aging is:
- DNA damage happens (In the nucleus and mitochondria)
- That leads to DNA mutations (which can later on become cancer) and epigenetic/chromatin alterations
- Cells have to decide whether to spend energy maintaining their chromatin or fixing their DNA damage. Indeed some of the proteins that help upkeep chromatin are also involved in repair (like sirtuins).
- This loss of chromatin stability means that regions that should be open are closed and those that should be close are open. The cell becomes less functional and resilient over time.
- Eventually an insult to the cell is stronger than its declining resiliency and dies.
Besides DNA damage, telomere shortening is the other variable I'd look at as a driver of aging, though we know, as will be discussed later, that one can fix telomere shortening and one still ages similarly.
Later in the post I will argue that all that has ever been done to extend the lifespan of organisms, from caloric restriction to reprogramming ultimately boils down to mostly fixing DNA damage or reducing its production.
Clocks and their weights: DunedinPACE
I found DunedinPACE quite intriguing beacuse it predicts a rate of aging, not an absolute number, and it is popular enough that there are people out there taking the test and publishing their own results eg here.
This below is from the DunedinPACE paper showing the correlation of the marker with age for their training set (The Dunedin cohort). There is some correlation but not that as strong as clocks that predict chronological age. This makes sense because DunedinPACE was built to predict how healthy someone is and that gets worse exponentially with chronological age, and so the rate of decline has to be greater later in life.
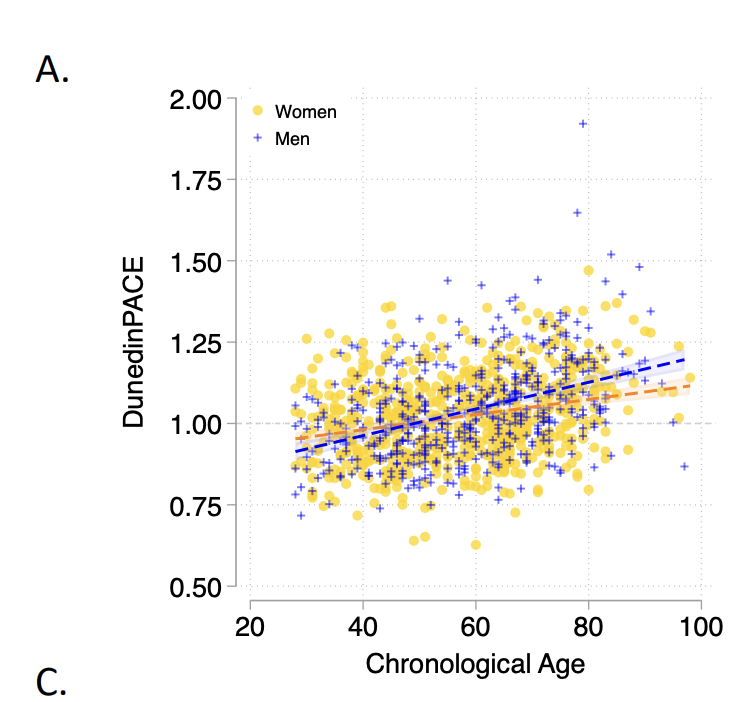
Now if you take this and apply it to two totally different datasets (The Normative Aging Study and the Framingham Offspring Cohort) one can ask the question: does a higher score in DunedinPACE predict higher mortality and morbidity? And the answer is yes it does:
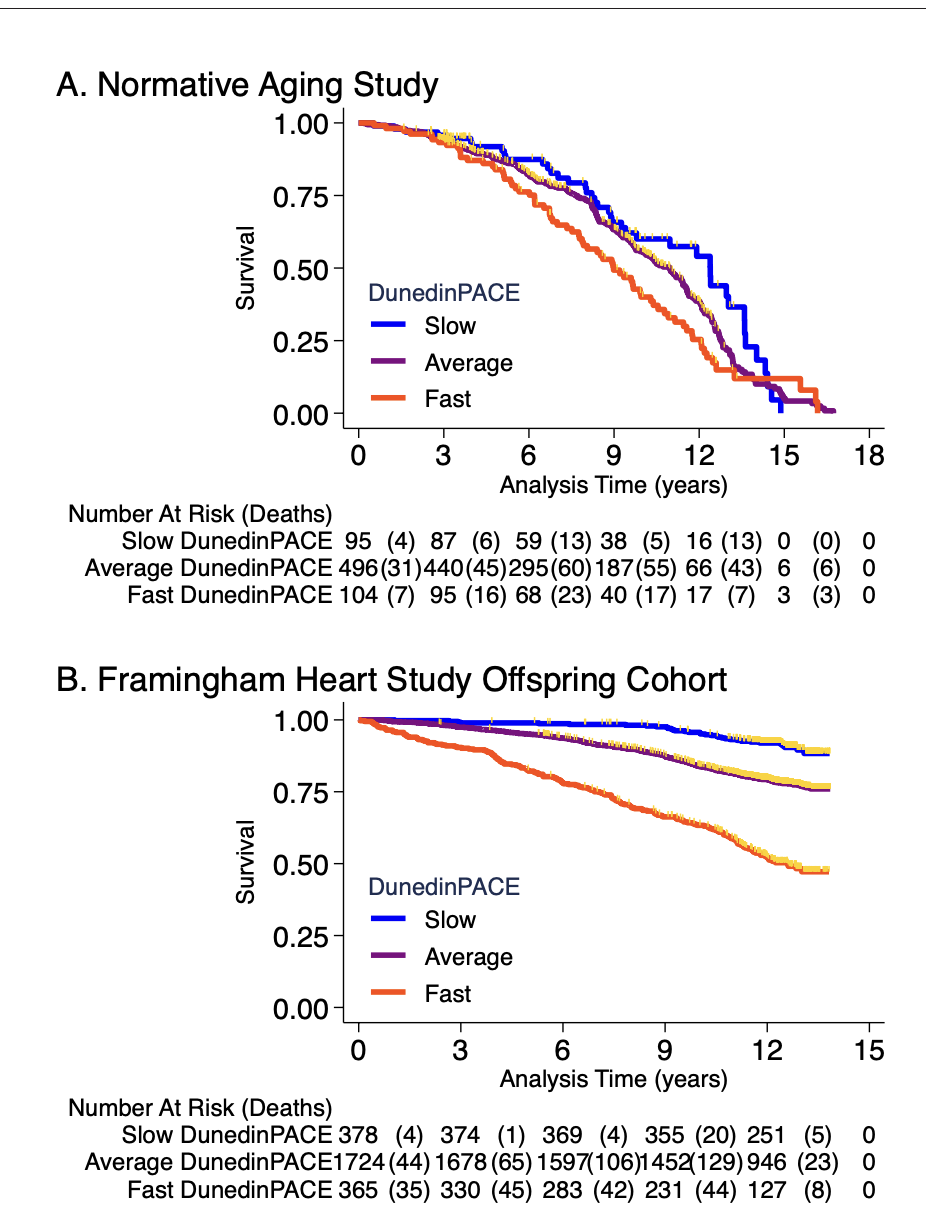
Framingham analysis included n = 2471 members of the Offspring cohort (54% women) with average age of 66 years (SD = 9) at DNA methylation measurement during 2005–2008. Over follow-up through 2018, 23% died, 13% were newly diagnosed with cardiovascular disease (CVD), and 6% had a first stroke or transient ischemic attack (TIA). Disability follow-up was conducted through 2015 based on participant reports of limitations to activities of daily living (ADLs) on the Nagi, Katz, and RosowBrelsau scales. Participants with faster DunedinPACE at baseline were at increased risk for CVD, stroke/TIA, and mortality (CVD HR = 1.39 [1.26–1.54]; stroke/TIA HR = 1.37 [1.19–1.58]; mortality HR = 1.65 95% CI [1.51–1.79], Figure 4B). They were also more likely to develop disability (Nagi ADL IRR = 1.40 [1.19–1.65]; Katz ADL IRR = 1.33 [1.16–1.53]; Rosow-Breslau ADL IRR = 1.39 [1.24–1.56]).
The average starting age in Normative aging was 77 years old and in Framingham, 66 years old. The effect sizes seem quite different and may have been affected by selection bias: In Normative Aging if you have already survived to be 77 years old you are probably healthier to start with so the clock struggles to tease out among a population of already healthy people whereas in Framingham the effect is clearer (and the sample size bigger).
In the supplement, they look at predictive power for mortality or quality of life (disability) after controlling for age. Controlling for smoking is also useful because smoking is an easy way to predict mortality that has little to do with age and we wouldn't want clocks just overindexing on this. This is quite important: there is no value if a clock is just predicting chronological age because we already have access to that. Unsurprisingly, DunedinPACE (and GrimAge, a clock built to predict mortality) do much better than clocks that try to predict just age:
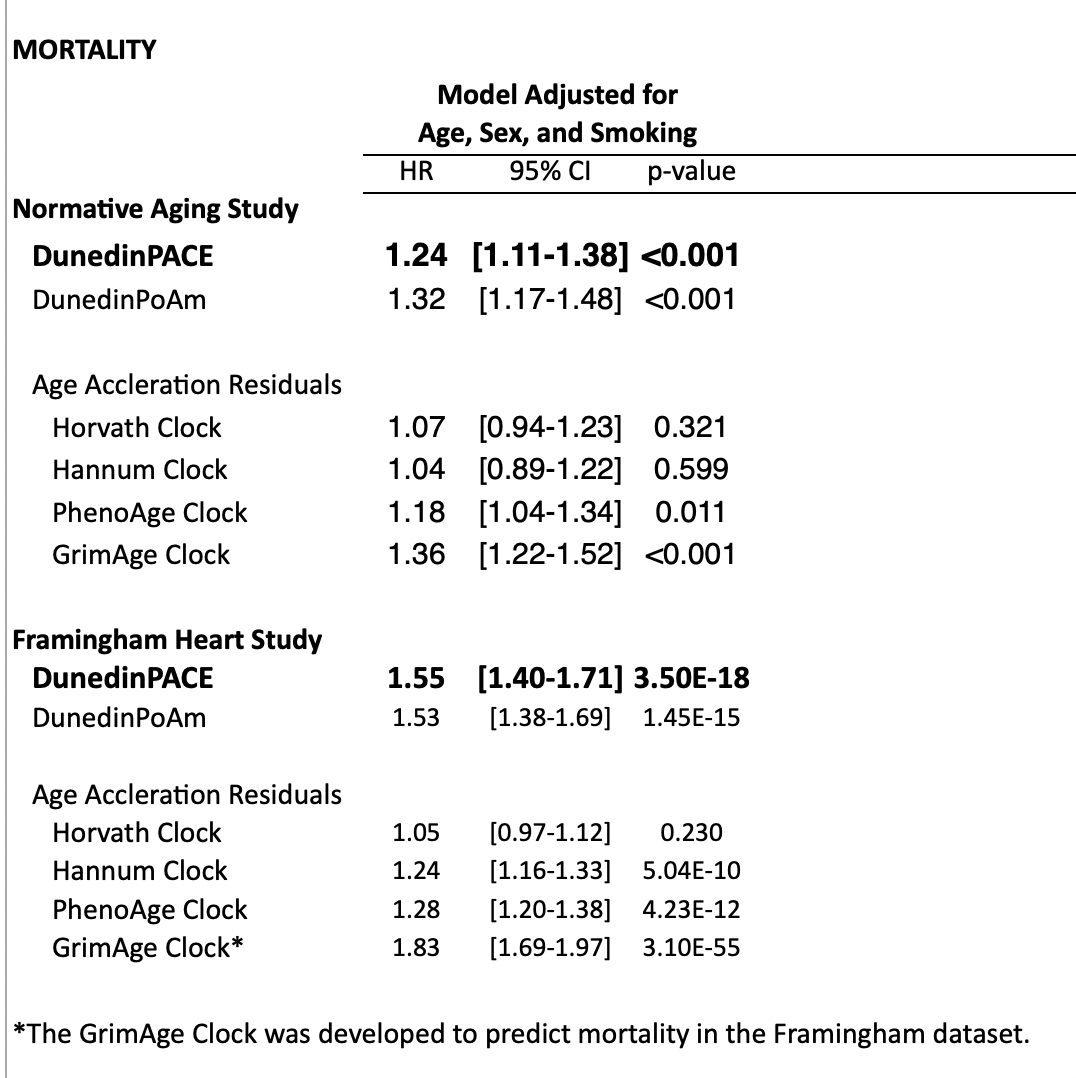
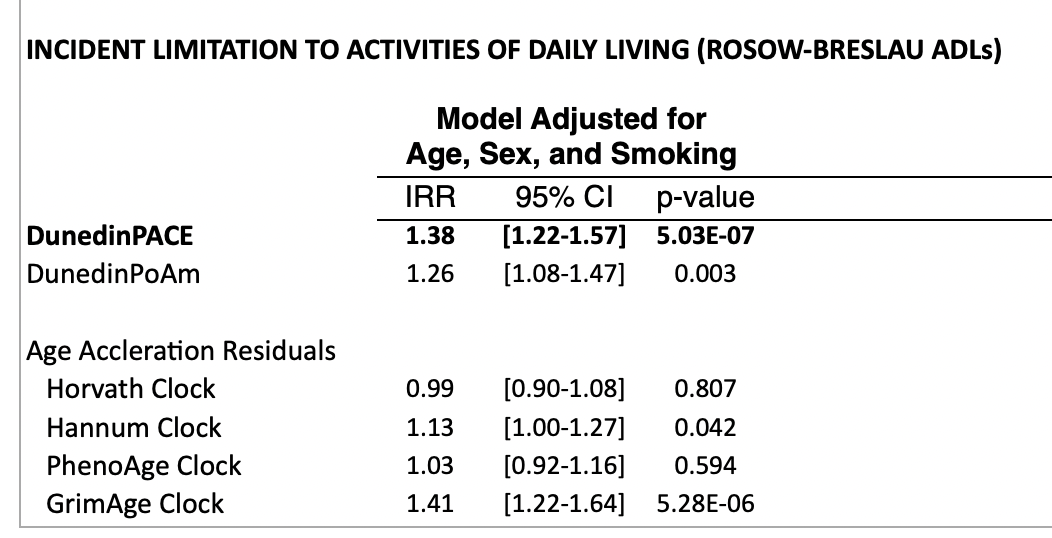
One might object to this that while these clocks work on a normal population, they might fail to detect out of distribution interventions. That is: If we were to subject a human (or a mouse) to caloric restriction or rapamycin or an intervention that turns on their FOXO3 (the one gene most associated with human longevity), would this show up in the clocks? Or, for that matter, if we just give people statins (which should lower LDL cholesterol), will that be reflected in the signal?
Mechanistically, one can investigate the methylation sites (CpGs) that the clock is picking up and see what these clocks might be picking up. This is rarely done!
I pulled the CpGs from their GitHub and slapped the Illumina annotations on top, then did some ChatGPT-assisted plotting. First, ouf of 173 CpGs in the clock, only a handful are quite large, followed by a long tail (Pareto principle at work!)
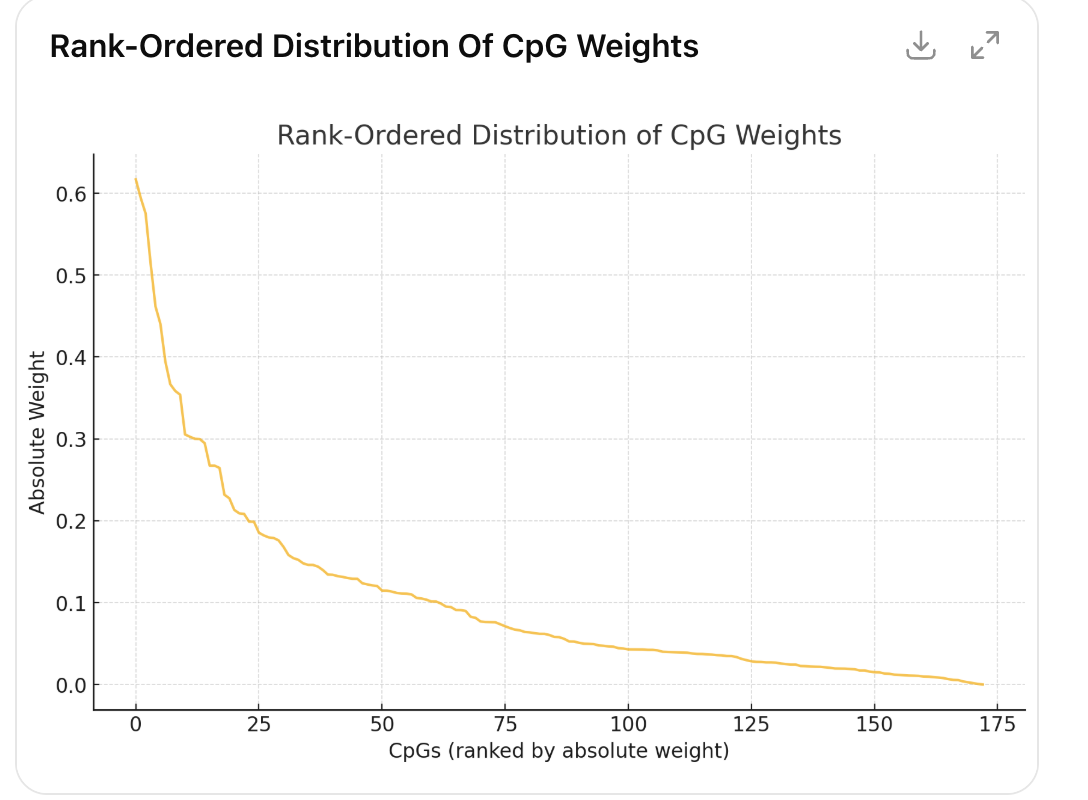
| probe | Gene Name | weight | Interpretation/Association |
|---|---|---|---|
| cg06570125 | — | 0.617 | ? |
| cg02650017 | PHOSPHO1 | -0.594 | Diabetes, CVD risk |
| cg06500161 | ABCG1 | 0.575 | Related to statin use in part (ie being on a statin makes you older in the clock!) |
| cg01554316 | GALNT2 | 0.515 | Lipids, metabolism |
| cg26470501 | BCL3 | -0.462 | NK-kB related (inflammation) |
| cg17460386 | FAIM3 | 0.440 | B-cell immune aging |
| cg17501210 | RPS6KA2 | -0.395 | MAP/ERK signaling |
| cg17018786 | DISP2 | 0.367 | |
| cg18181703 | SOCS3 | -0.359 | Inflammation |
| cg15192750 | — | 0.354 | |
| cg05304729 | MNDA | 0.305 | Inflammation |
| cg13274938 | RARA | 0.303 | Inflammation/immune |
| cg10919522 | C14orf43 | -0.300 | Smoking? |
| cg27165794 | PNMA1 | -0.300 | Cancer? |
| cg14702960 | — | -0.295 | |
| cg16924010 | — | 0.267 | |
| cg17439800 | — | 0.267 | Obesity? |
| cg00574958 | CPT1A | -0.265 | Metabolic syndrome |
| cg01055871 | EHD2 | -0.232 | |
| cg09349128 | — | -0.227 |
But are these causal? You can already see that one of the top driving CpGs is known to be driven by statin use. People on statins are plausibly less healthy but the statins are not what is making them sick. So were someone to go off a statin, their DunedinPACE score would show rejuvenation but their mortality risk would increase.
This is why it is important to look at how these clocks are constructed and investigate whether the CpGs have reasons to be causal or not.
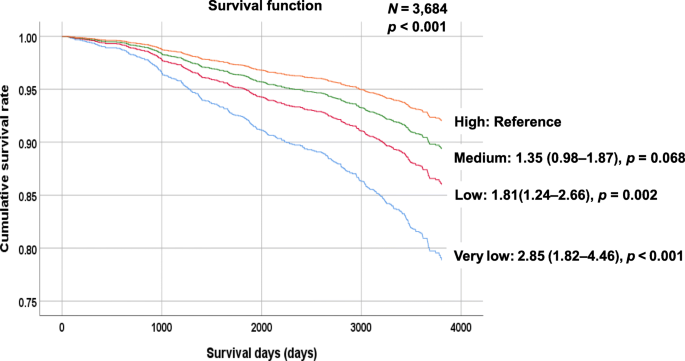
It is also useful, I think, to present stratified mortality curves according to other biomarkers just to illustrate that mortality prediction is not anything magical that only clocks can do.
LDL cholesterol
[source]
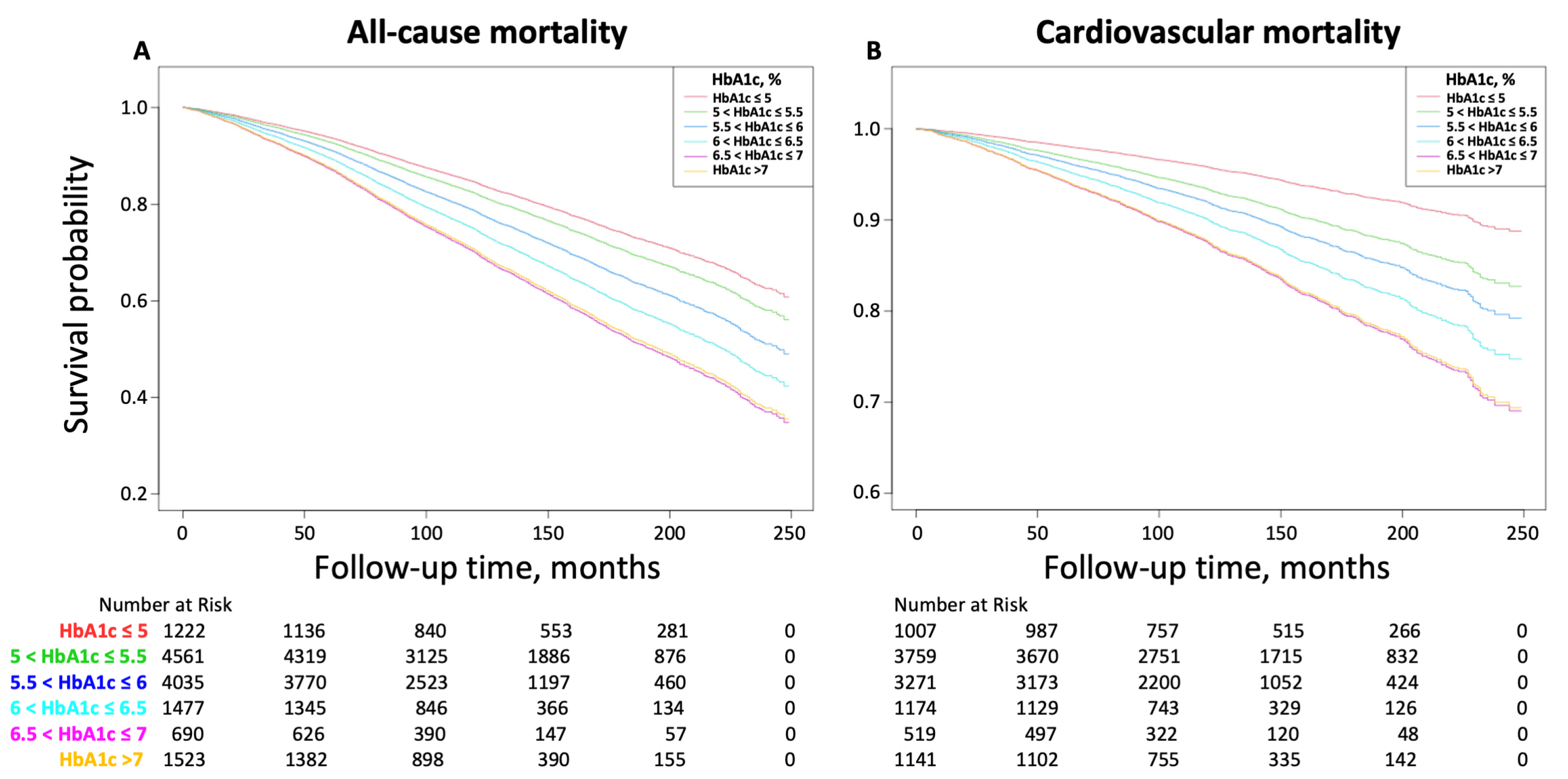
Glycated hemoglobin (Hb1Ac)
[source]
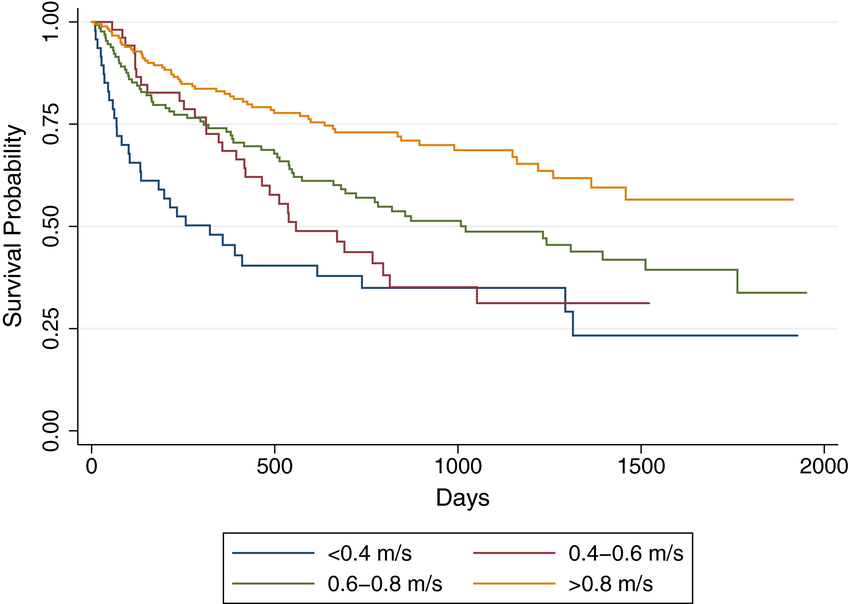
Walking speed
[source]
Physical strength
[source; in this plot maximum quadriceps torque is depicted]
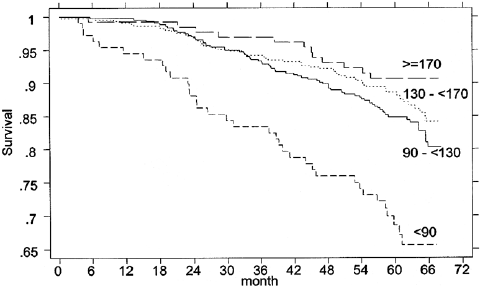
Can you lift you way to immortality? The search for causal aging markers
These examples are illustrative of the way the causality of disease and aging work: aging causes disease, and disease causes mortality and disability. As a result, disease is causally closer to what we observe (people dying and suffering) so if one tries to make predictors of those one will pick up stronger drivers or correlates of that than of the underlying driving factor (aging).
And because in the natural population the decline of everything is correlated to an extent, one can use one (grip or leg strength) to predict another (mortality) despite the fact there is a very small direct causal link between having really strong forearms and dying; rather strenght acts there as a proxy for overall health.
This is not the case for causal biomarkers like LDL cholesterol which cause the relevant disease (such that modifying it modifies the disease) and we can measure directly.
Hence we can measure the downstream consequences of aging very easily and we can predict outcomes of interest reasonably well, but that can all be done without touching the measurement of cellular aging at all. We don't even need these clocks to do that!
Suppose you could either know your DunedinPACE score or know your Hb1ac (diabetes risk), LDL/ApoB (CVD risk), your lean mass %, and the usual blood counts and metabolic panels? What would give you a better picture of your health and mortality risk. To me, it is uncontroversially clear that classic validated biomarkers are not only superior predictors but they are also directly actionable compared to epigenetic clocks. If your LDL is high you can take statins. If your DunedinPACE score is high what are you supposed to do?
To my knowledge, no analysis has found that methylation clocks are superior to composites of traditional blood based biomarkers to predict mortality and disability. If you are aware of any such analysis, let me know!
But if we have these blood-based biomarkers that work so well, what's missing? What's missing is models that estimate aging, instead of those two other downstream things.
Sure, but who cares about aging you might ask? We care about its downstream consequences right? If an aging predictor cannot predict mortality better than LDL cholesterol, what would be their use?
Good question! One that doesn't get asked often enough. Moreover, is aging even a thing you can measure with a single number? Right now we don't measure "health", we measure diabetes risk and CVD risk separately.
One answer could be "for research purposes". If we can slow down the rate of aging permanently in a mouse, then we can know that very quickly whereas waiting for a cohort of mice to die and/or get frail can be expensive and time-consuming.
Another use could be for clinical trials, if one shows age slowdown in mice and wants to take that to the clinic, one could use such a biomarker to have an early readout of whether the treatment is working or not and if its not, discontinue and save money on the trial.
In the appendix to this essay I discuss proteomics clocks, which I argue are superior to methylation clocks, and superior (and deeper) to the simple baselines of LDL+HbA1c. They do something methylation clocks struggle with: sampling different tissues with specificity; methylation clocks tell us the methylation status of a given cell type (blood cells), but not say how the brain is aging. But there are plasma-based biomarkers of CNS disorders like Alzheimer's: the organs leak information about their state into the blood, and that can be read out with proteomics. So I argue that in the future, paired proteomics clocks (for intantaneous organ-specific function) with methylation clocks (aging of blood cells) will give us a reasonable picture of how a living being is aging much better than what we currently have right now.
But now, back to the methylation clocks! We said the issue is that if we train them on endpoints like mortality, we bias them towards disease, not aging. What can we do about that?
Doing (aging prediction) by non-doing (aging prediction)
There are a couple of options to work around the limitations of the data we have with methylation clocks
-
Correct the mortality/disease signal by broadening or standardizing the population:
-
Try to include other species in the dataset so that it picks up what is common across them and not just in humans (diabetes, etc)
-
Develop clocks for populations with standardized environments and genetics (like lab mice or cells in culture!) to remove the effects of specific diseases from biasing what the clock picks up
-
Leverage EWAS to try to pick up CpGs that are associated with specific diseases
-
-
With a theory of aging at hand that points to drivers of aging:
- Hand-pick CpG sites that are posited to be drivers of aging as opposed to letting a model optimize the weights (One man's taste is another's bias).
For the first approach there we can look at the CpGs of this other clock (PanTissue, PanMammalian). But this is not great: It doesn't uniquely pick out the genuine slower rate of aging of dwarf mice (thought they did claim this in the preprint and then took it back!), which by all metrics age slower, nor does it pick out the substantial epigenetic rejuvenation of iPSCs. So perhaps adding all species is bad and adds noise.
What about clocks just trained on human data, and then applied in vitro? That is more promising: these clocks show that the cells age in culture in a linear fashion (just like humans in the wild), just 60-65x faster.
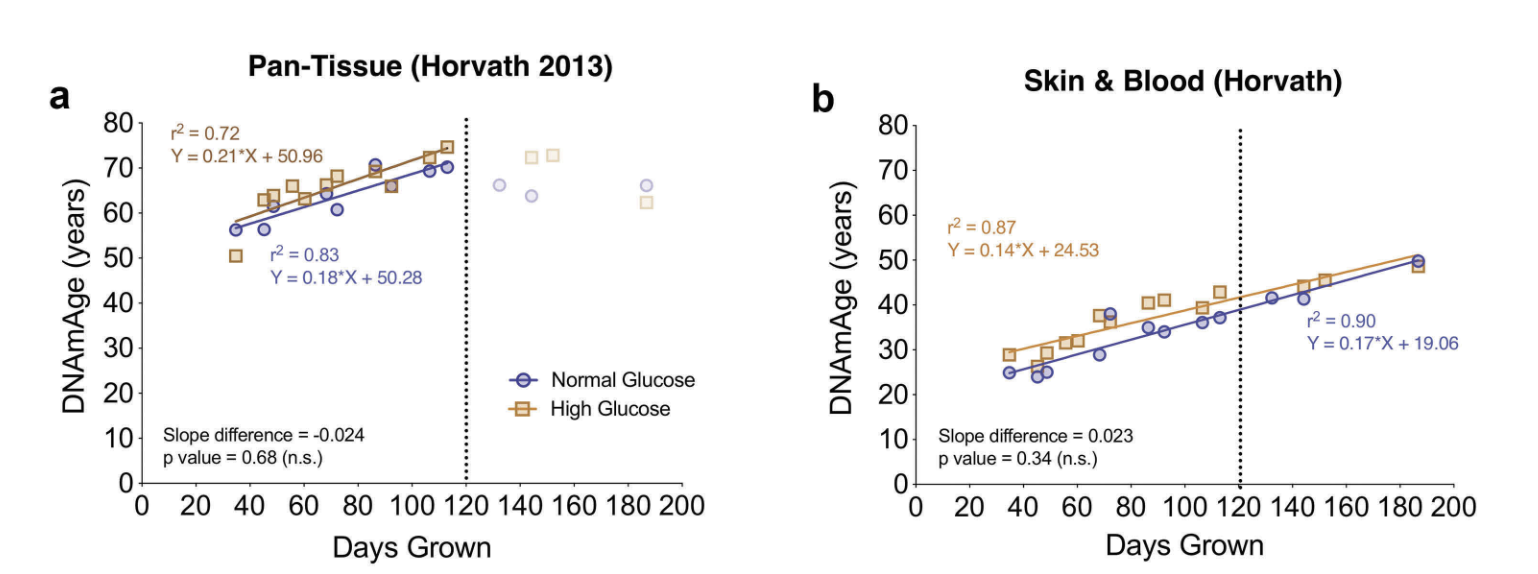
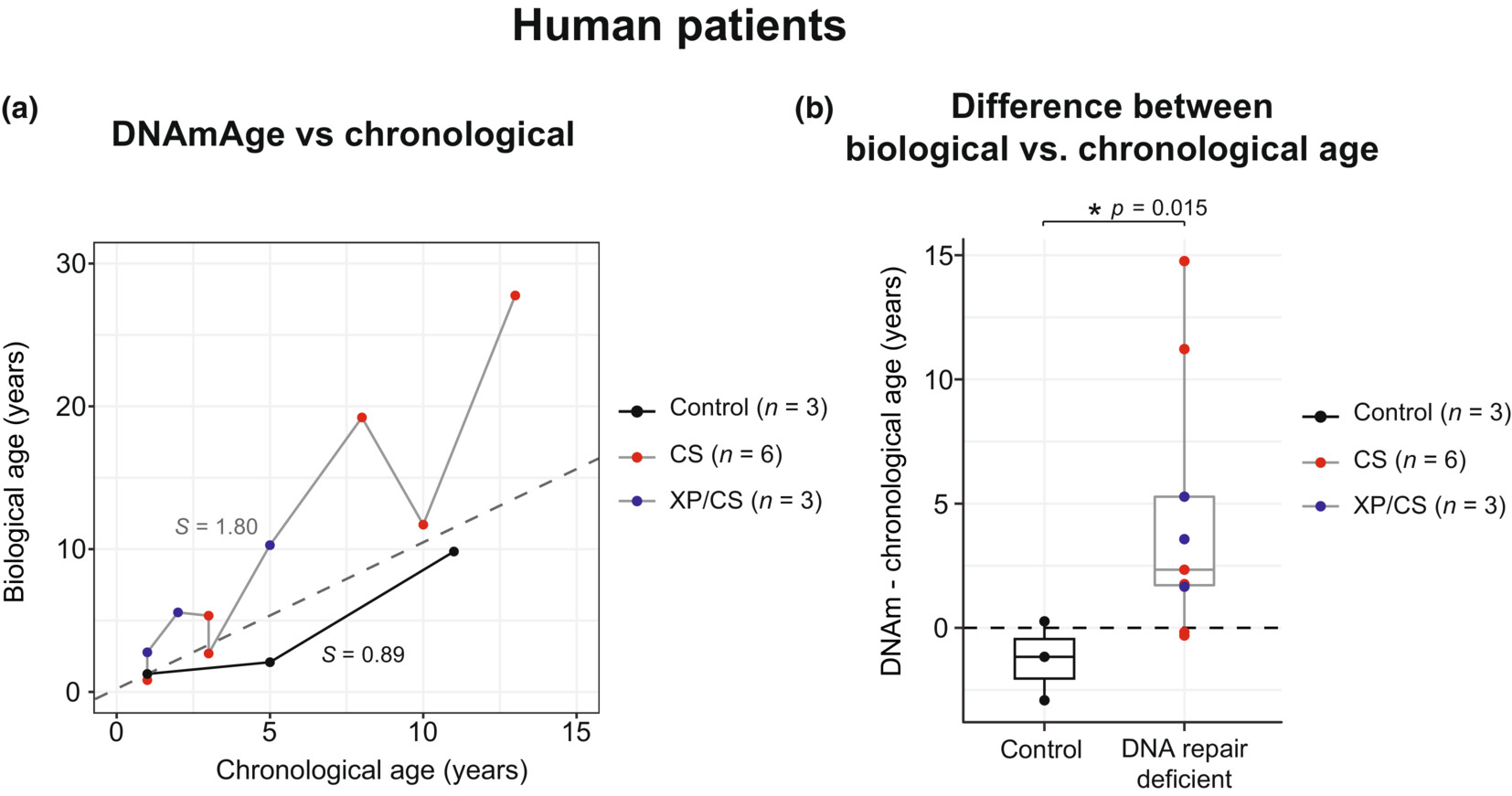
The Pan-tissue clock also seems to work in patients with certain progerias (accelerated aging)
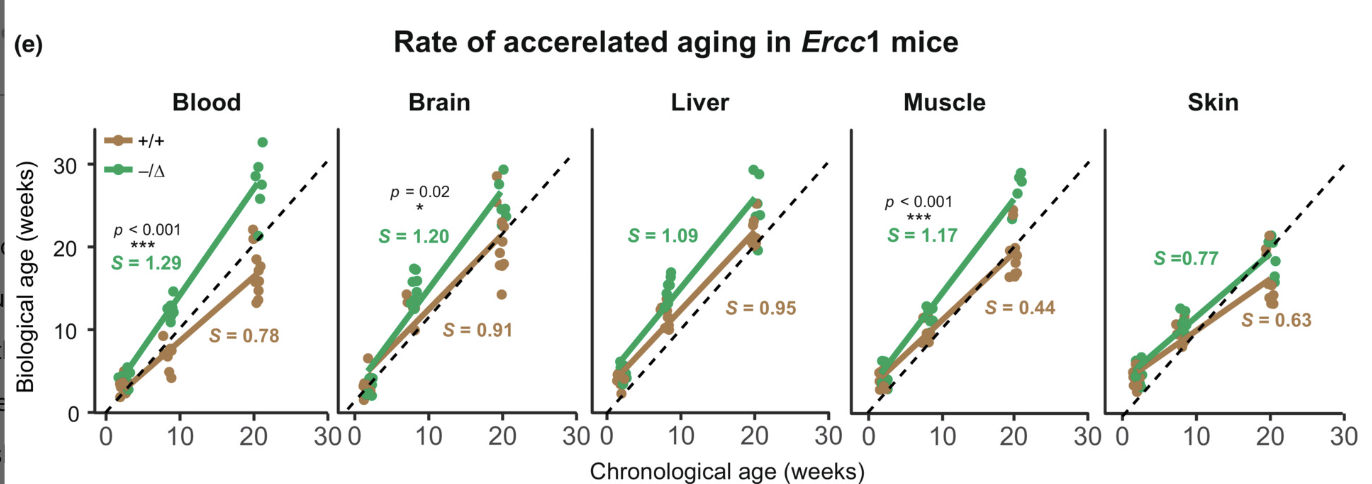
And it also performs well for Ercc1- (a DNA repair gene defect) mice (Where genetic and environmental variablity is controlled), at least for some tissues even though this clock was trained on humans not mice.
Retrotransposon clocks
Another approach to make better clocks is to handpick CpGs. A good candidate for this are CpGs that suppress retrotransposons. Retrotransposons cause DNA damage when they insert themselves in DNA and they get derepressed with aging. One can build a clock that looks at CpGs that are in Retrotransposons exclusively:
Intriguingly the authors note
Notably, we observed ourRetroelement-Age V2 clock overlapped with 9 CpGs (cg06672696,cg07286682, cg08822136, cg16936289, cg16810279, cg22277154, cg13261390, cg22277154, and cg24251135) used in AdaptAge,CausAge, and DamAge causality-enriched epigenetic clocks recently developed using Mendelian randomization (Ying et al., 2024). This observation suggest some of the signal from our Retroelement-Age V2 may include sites that contribute and/or protect against aging.
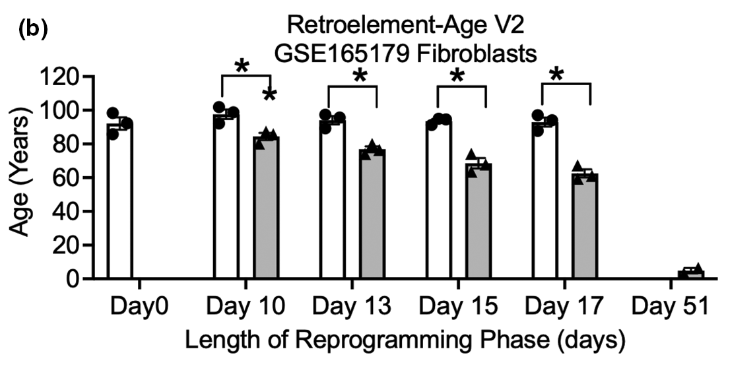
This works for reprogramming:
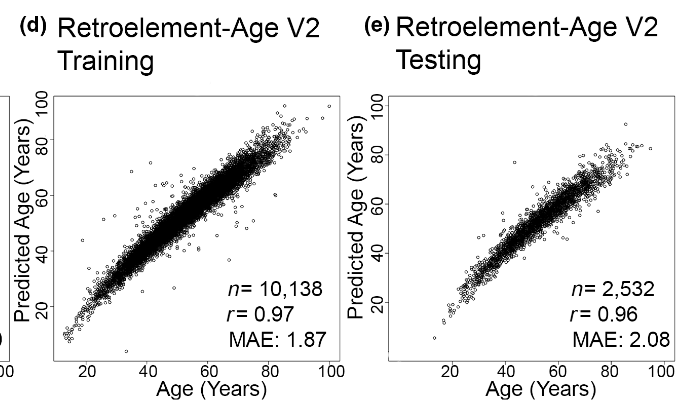
It also predict aging in vitro:
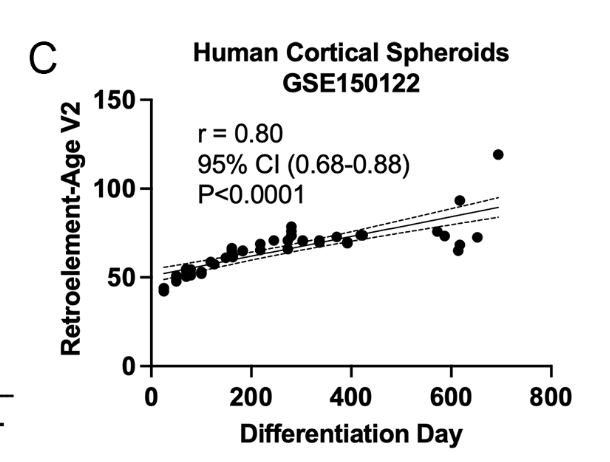
Alas, this clock hasn't been applied to other datasets in eg slow aging mice or mice subjected to caloric restriction. A similar approach, however, has, and that shows the expected effects:
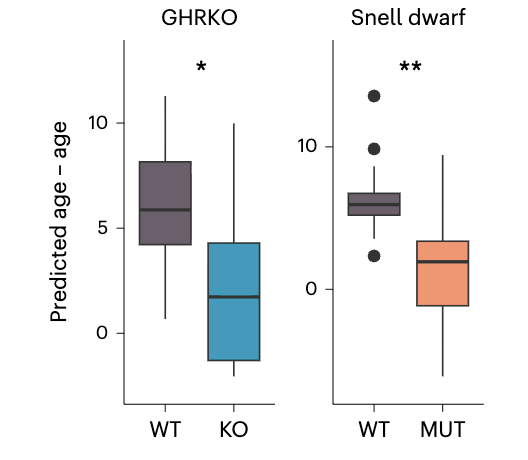
For context, these Snell dwarf are a well known case of slow aging:
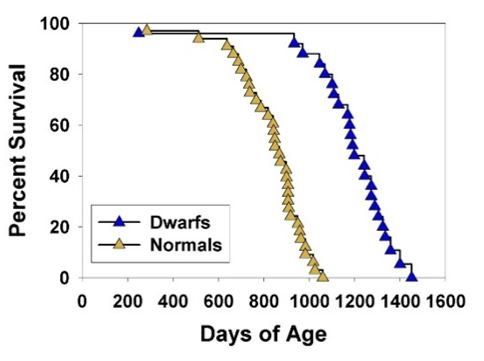
It also seems to match with the idea that bigger dogs age faster (bigger dogs have their retrotransposons derepressed faster):
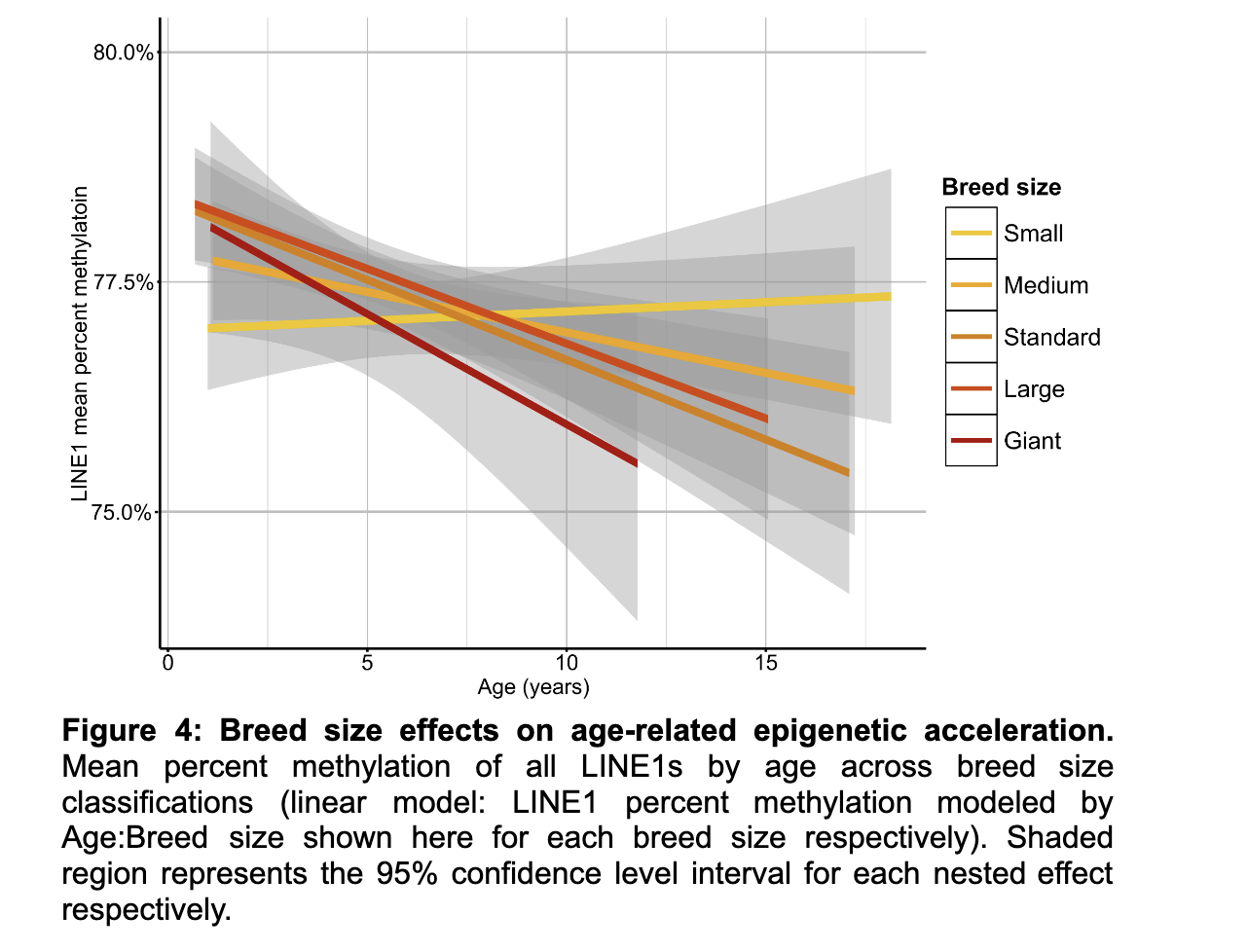
I really like the retrotransposon clocks! They do seem to be neatly tied to DNA damage. With these, I think we have some grounds to believe, given what we know about retrotransposons, that if one were to see a slowdown in this clock, one would have thus slowed down aging. But we could not be maximally confident: If someone developed an epigenetic editor to methylate all these retrotransposons, that could drop the Retrotransposon age to zero, and yet that wouldn't necessarily reset aging as it is not the only source of DNA damage. Using this clock would be valid especially if one's not specifically targeting retrotransposons, but even then I would suggest using a panel of approaches and not just this one.
Aging clocks with mouse-only data
Other clocks, developed specifically for mice are also able to pick this up, doing it better (greater separation) than what the Retrotransposon-only clock does:
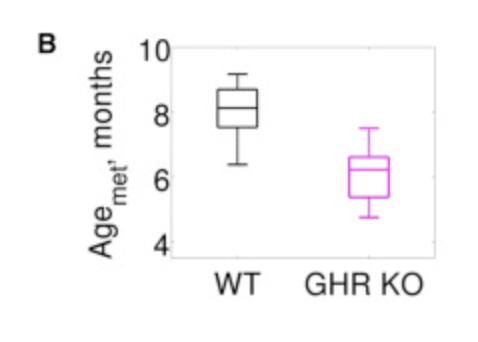
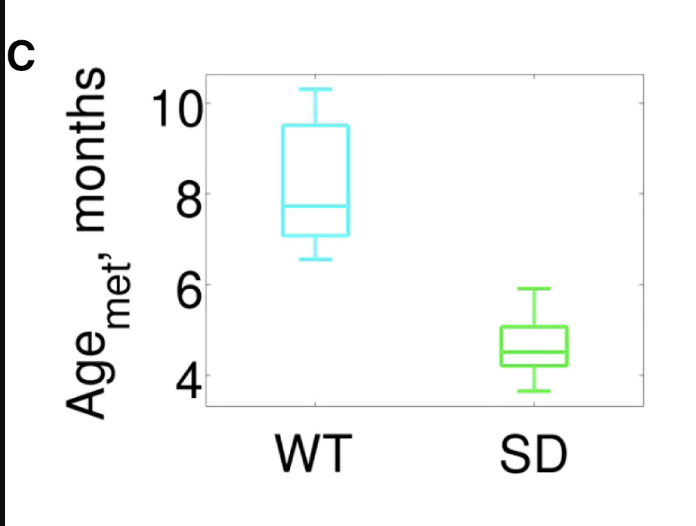
And later this one as well, also from the Gladyshev lab.
Aging clocks through EWAS
More recently, we got this paper from the Gladyshev lab that I really liked trying to build "causal clocks". The idea here is to carefully tease out what CpGs might be associated with pro-aging vs those that are differentially altered with chronological aging as a response to aging. The fist one they call DamAge clock, the second, AdaptAge. DamAge passses the reprogramming test (age drops with reprogramming):
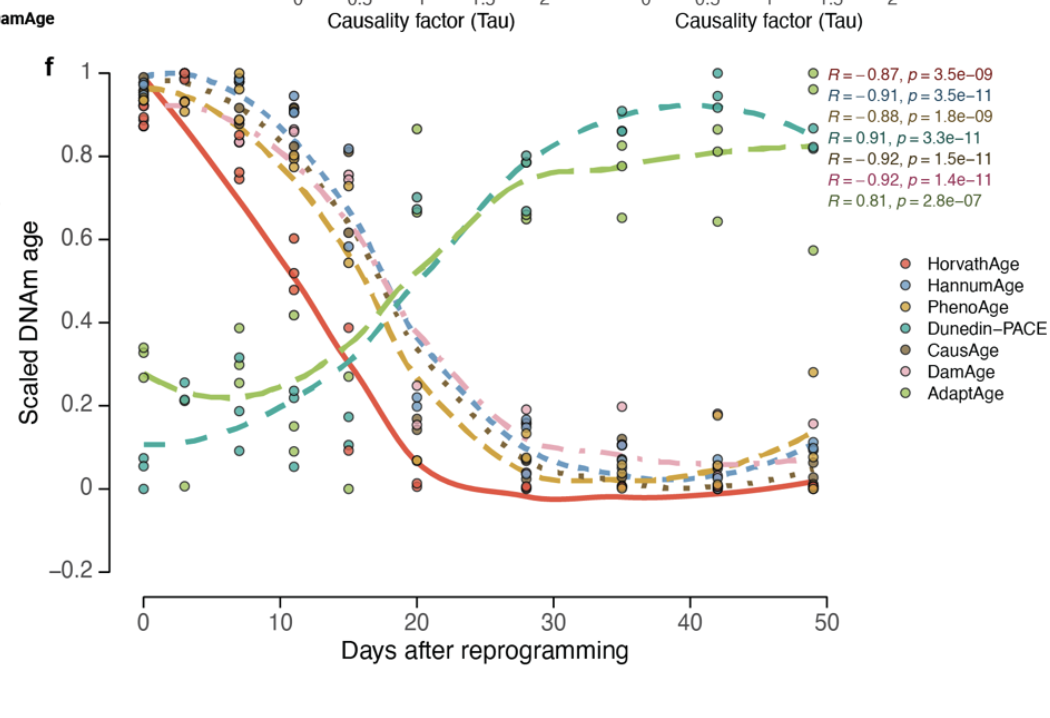
It picks out progeroid damage, exposure to cigarette smoke, and exposure to UV light (ie skin samples exposed to the sun vs non exposed)
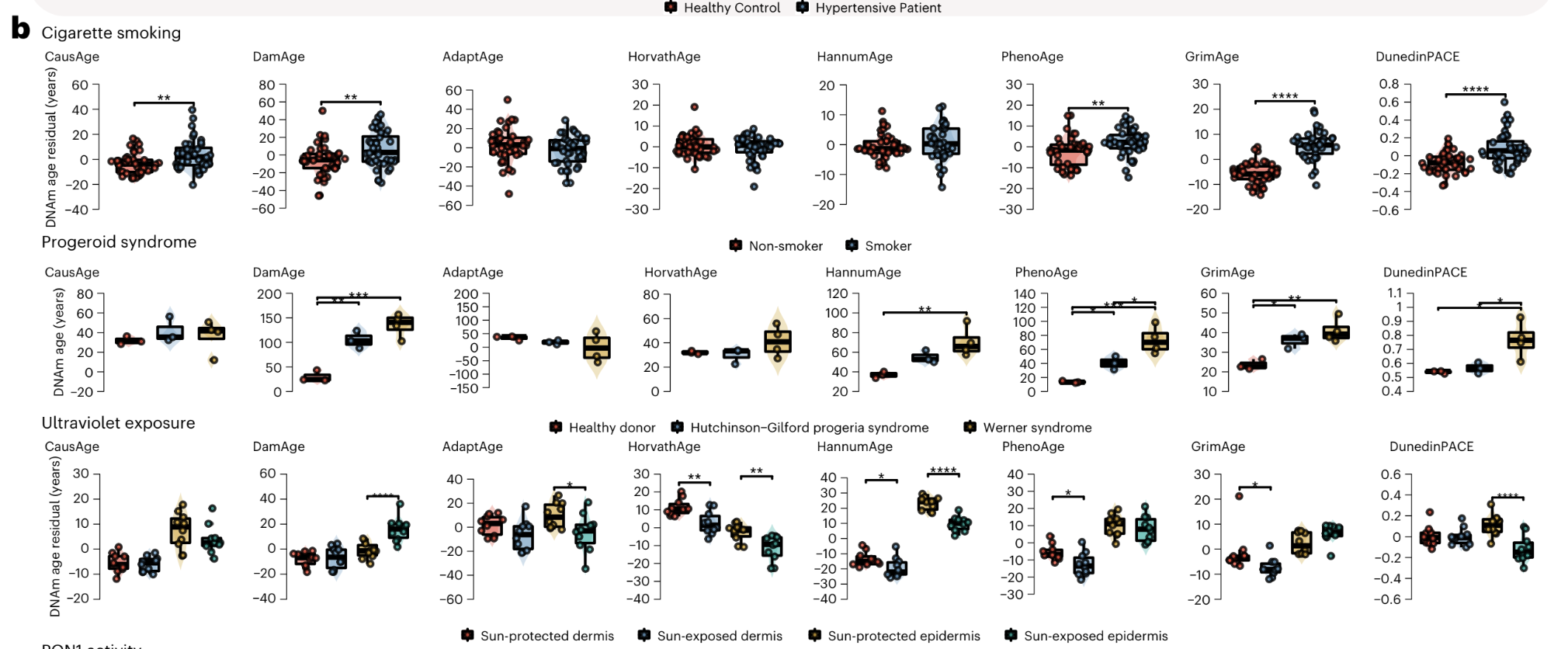
Here the other clocks fail to pick this up, in fact some say UV exposure slow down aging! But no, what they are probably picking up is mechanisms that counteract aging in those cells. DamAge correctly assigns a score similar or greater to these exposed cells.
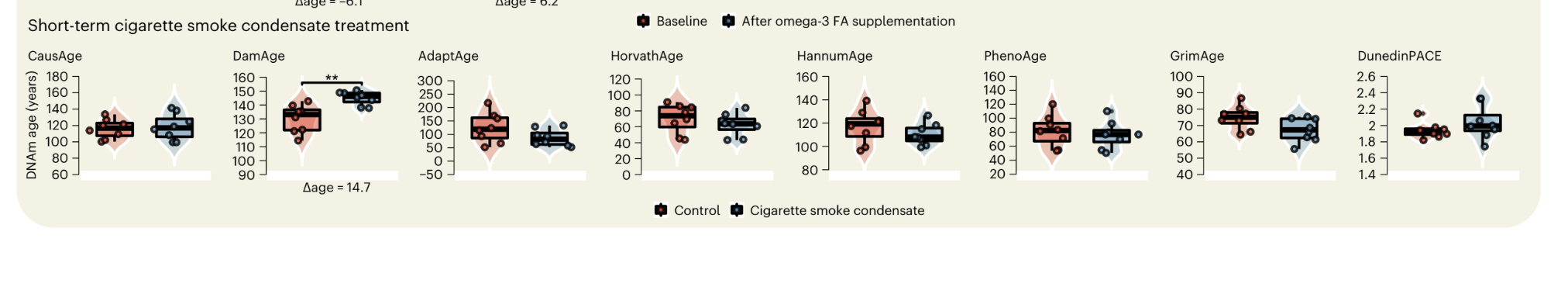
In vitro, the effects are more stark, exposing cells to cigarette smoke shows up cleanly only in this clock:
Intriguing. So what is this clock picking up? The weights are here. Doing the same as before, here's some ChatGPT interpretation.
First, most of the weights of the model are negative: this means the CpGs picked up are to be interpreted as "the more methylated the gene is (the less expressed), the lower the damage"
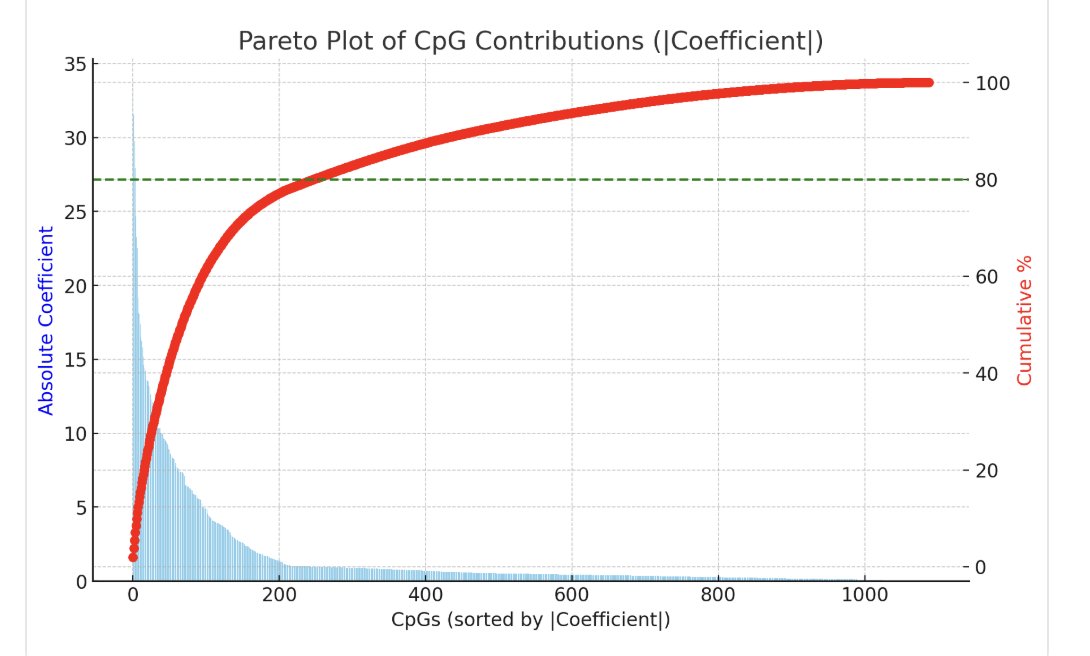
These are the top 20 CpGs
| CpGmarker | CoefficientTraining | UCSC_RefGene_Name | Interpretation |
|---|---|---|---|
| cg07850154 | -33.64986406 | RNF180 | Also here, involved in proteostasis. The entire gene seems to get less methylated over time. |
| cg02254885 | -31.56205228 | EFCAB3 | |
| cg05463027 | -29.7405477 | KIF13A | |
| cg08529529 | -27.93679343 | ALOX5AP | Also here |
| cg08526814 | -24.67192062 | DNTTIP2 | |
| cg07495704 | -23.24721626 | ||
| cg02867102 | -22.53153968 | Also in GrimAge, smoking | |
| cg04229059 | -19.17402516 | SLC38A7 | |
| cg22652782 | -18.1396963 | MYBPC2 | |
| cg01082242 | -18.03942941 | HIF1AN | Inhibitor of HIF1a (hypoxia inducible factor). |
| cg08593364 | -17.42228852 | SLC35F2 | |
| cg08081725 | -16.50151564 | NDE1 | |
| cg23282585 | -16.24094824 | HIST1H3G | Histone H3.1 (replication-dependent). More cell replication? Picking up cancer? |
| cg09754948 | -15.80284482 | ATXN2L | |
| cg26635214 | -15.1806246 | HSD3B7 | 3beta-hydryxosteroid dehydrogenase type 7: bile acid synthesis. |
| cg15063695 | -14.61865262 | GK2 | |
| cg03950166 | -14.36951799 | FAM108C1 | |
| cg01557754 | -14.19144026 | FGF11 | |
| cg02339392 | -13.60087486 | ZNF187 | Associated with CKD |
| cg03520471 | -13.57242019 | GABRR3 |
Then here's pooled by gene
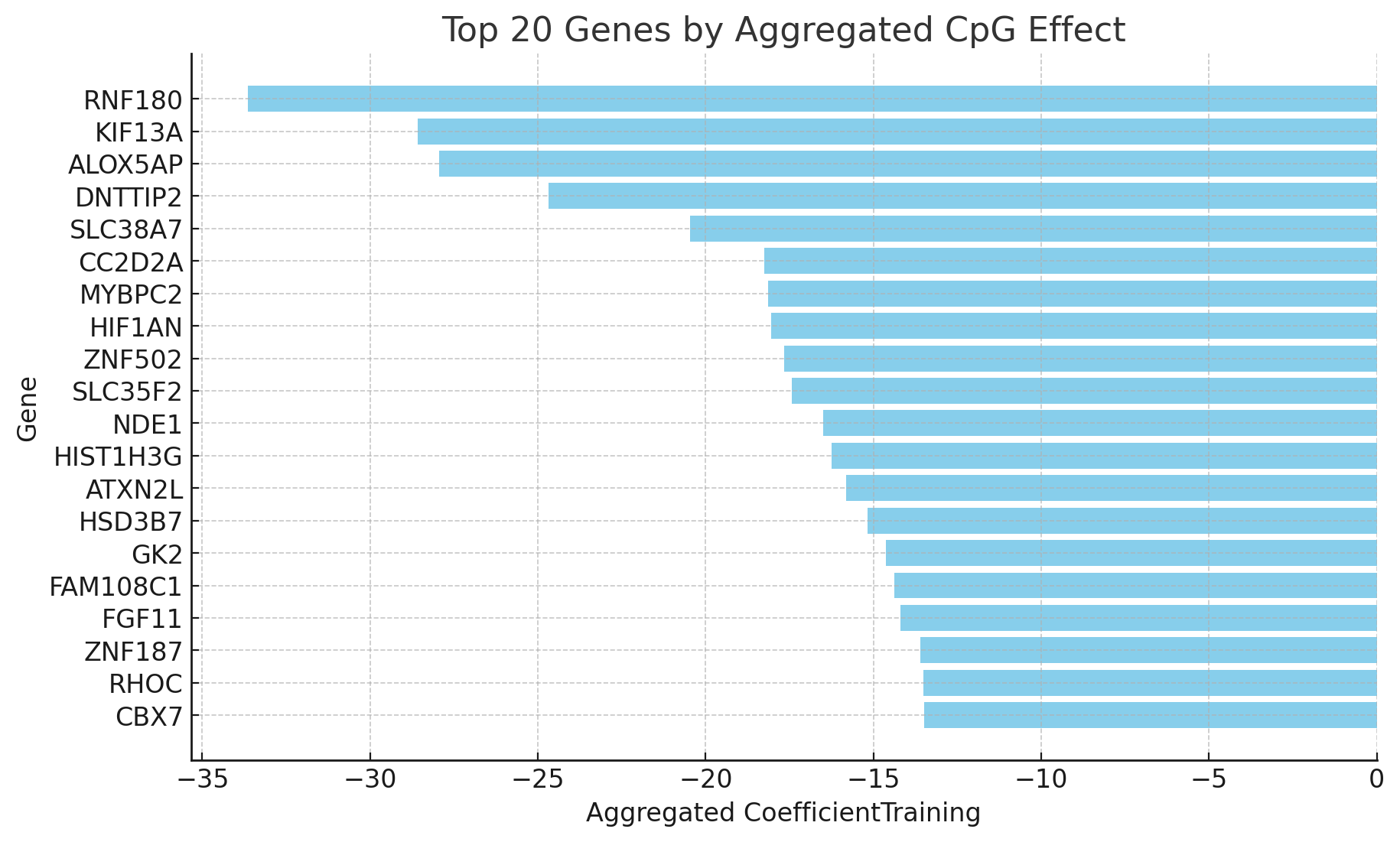
This looks much better than DunedinPACE: the CpGs and genes picked up are not obviously tied to diabetes and metabolic syndrome! But it's still hard to interpret.
Aging clocks with in vitro data: Polycomb Repressive Complex sites
One last way I'll discuss of constructing more robust models to track cellular aging is training them in vitro where organismal-level disorders are not relevant. This, as it turns out also has its problems.
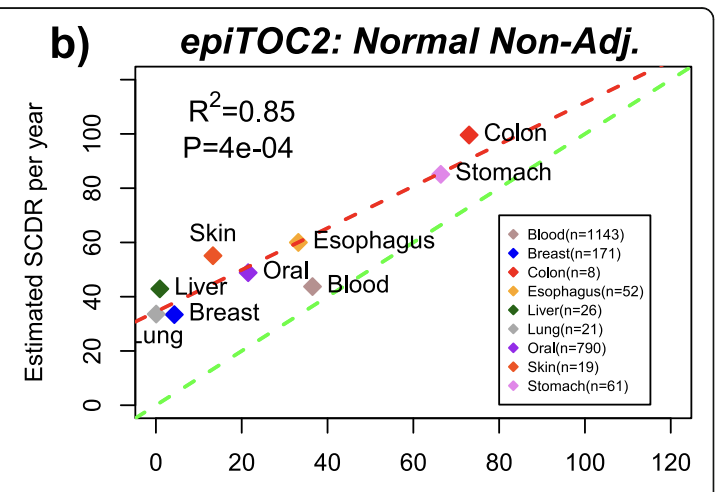
The EpiTOC/pcgtAge clock (Yang et al. 2016) was constructed by handpicking as opposed to running a regression which makes it more robust to allegations that it's picking just a correlation. What they do is pick PRC2-related promoters that get hypermethylated with age in blood samples and that start with no methylation across a range of fetal tissues, then the score for a given sample is simply the average methylation across those CpGs. This can be used to predict human chronological age, and it also correlates with the number of cell divisions estimated for stem cells in different tissues, which matches also with other clocks that pick up tissues that turn over faster as faster aging. Note that in this picture below each point is a different tissue from a person of a different age.
A later model, EpiTOC2 (Teschendorff 2020) further expands this methodology, using fewer CpGs.
The original Horvath clock has 353 CpGs that are jointly fitted to estimate the chronological age of a sample. This is not great because
a) Plausibly some people might have aged faster or slower but we are telling the model that two samples have an age of exactly their chronological age
b) Different tissues plausibly have aged faster and slower but we again tell the model to assume that they all have the same age
EpiTOC2 instead models each CpG individually, they say that the methylation at a site equals is a function of someone's age, how often stem cells divide per tissue, and how likely is that a cell division will lead to an increase in methylation. Doing this they fit one model per CpG (3 values to fit), then pick the modal value of the stem cell division rates estimated (should be the same across CpGs because they are all replicated jointly) and fit the model again, this time with just 2 values (de novo methylation probability and the ground state methylation).
Though this all is done in blood, their model is posited as universal per cell division so then this model fitted in blood can be applied to other tissues. So given say a colon sample, one can measure the actual methylation and knowing the age of the person one can calculate the number of stem cell divisions for a tissue. They find their estimate of this parameters correlates reasonably well with literature estimates of how often stem cells divide per tissue
Moqri et al. (2024) did something similar in concept (picking PRC2-associated sites and averaging their methylation) but without modeling, they just find those PRC2 sites, pick 1000 with the most PRC2 binding and average the methylation of that.
This, they explicitly note, is what explains ELOVL2 (discussed later): It is one of those PRC2 associated sites!
Exciting, isn't it! But: Is this causal? If we were able to revert this clock while leaving everything else unchanged, would we revert aging? I don't think so!
I looked into why is it that cells keep adding methylation at these sites and the reason seems to be that initially those genes are silenced during development when cells are comitted to a fate, by depositing H3K27me3 (a histone mark) on it. But during cell division, this gets diluted and to compensate, the cell methylates those sites to continue to repress them (Yang et al. 2023). This means that these PRC2 methylation sites are non causal! They are an adaptive response. The implication of this is that if we were to remove them without adding back H3K27me3 we would make the cells younger "by the clock" but make them less functional. Conversely, this means it is also possible to make the cells genuinely younger while seeing no change in an epigenetic clock that heavily indexes on these sites. It also means that the sometimes reported age acceleration in cancer might just litearlly be a proxy for their higher rate of cell divisions (Rozenblit et al. 2022).
Everything is connected: epigenetic clocks are not unique, but they are convenient
The state of a cell is the aggregate of everything that's happening in the cell: how many mRNAs of each type there are, concentrations of proteins, what chromatin is closed, etc.
But it's all connected: One influences the rest. As a result, if one measures deep enough a cell, one can reconstruct the rest of the state from that subset. From the genome and chromatin one can predict gene expression, and from gene expression one can predict protein abundance and chromatin accessibility3 .
If one believes this then there's the question of "how deep to sample" and "what sorts of things can be sampled". The genome is not that useful in general for aging because cells can mostly work around the damage that the genome is subject to, though one can predict aging, albeit noisily from somatic mutation accumulation (Alexandrov et al. 2015; Koch et al. 2025), one would not be able to predict say cell type just from the genome whereas this is possible from the other, more dynamic -omes.
Single cell RNA seq is something people have tried to make clocks out of (Buckley et al. 2022) but generally those approaches rely on pseudobulking cells to reduce the noise of mRNA expression. Bulk RNA clocks work much better. But still they seem to be noisier than methylation clocks or, for that matter, bulk ATAC clocks. Chromatin is more stable than mRNAs and so a better thing to measure to get a consistent readout of state unbiased by transient effects.
The full proteome, as of today, is more expensive to measure, and we can't quite fully measure all of it, but if we were able to, it would probably also work fine to build aging clocks out of.
That makes methylation (or chromatin, even better) as the most reasonable way to make and use aging clocks.
Epigenetic clocks and organismal aging
When someone says that doing something changed their epigenetic age what that really means is the same as when someone typically makes a claim about their telomeres having lenghtened. It usually means the epigenetic age of the cells in the blood. That epigenetic age can change for various reasons:
- Change in cell type composition (which now some clocks try to correct for)
- Death of older cells "Rejuvenation by depletion (as in senolytics)"
- Replenishment from younger cells; this would have to come from rejuvenated hematopoietic stem cells in the case of blood.
- Change in state towards less inflammation. As in most populations older people have more inflammation, a reduction in inflammation would register as reduced aging.
- This also explains why what the clocks measure seem to increase and then decrease during disease (Poganik et al. 2023)
Additionally the readout of a clock can be biased by the source population in the training set. If you take say all Americans and build an aging clock on that and then a person that's fairly healthy does it, it is likely that they will show up a bit younger in the clock and that might be because of less inflammation.
That said, if someone has a substantial (say >10 years) reduction in their own personal score, I would take that as evidence of robust rejuvenation for cells in their blood. This has been achieved, to my knowledge, only through bone marrow transplantation, where an old person gets bone marrow from a younger donor; in that case the old person ends up with a real legit rejuvenated blood system (Soraas et al. 2019)4
These people have a younger epigenetic age and do live longer (DeZern et al. 2021), but they do not live nearly as long as the drastic reduction in epigenetic age would imply. That is, you can have "20 year old blood" in a "80 years body" and clocks would totally fail to register that the rest of the body is not quite 20.
Do neurons age?
It seems one of the most stressful things a cell can do is divide: tissues with more cell division age faster. This then leads us to the question: what about tissues where the cells don't divide much like the brain? Do those age very very slowly?
If one takes DNA damage as the prime mover in the aging process, one can look at kinds of DNA damage in DNA mutations: we have more access to that kind of data than we have for DNA methylation because people have been extracting tissue samples from all sorts of tissues for cancer research. This data has been analyzed for patterns in mutations and some emerged that seemed to correlate fairly well with chronological aging (Alexandrov et al. 2015). The two biggest "clocklike" signatures are SBS1 and SBS5 (Spisak et al. 2024), which together represent half of the mutations one will accrue with aging. SBS1 seems to nicely track cell division rates as it is flat in neurons and much accelerated in colon.
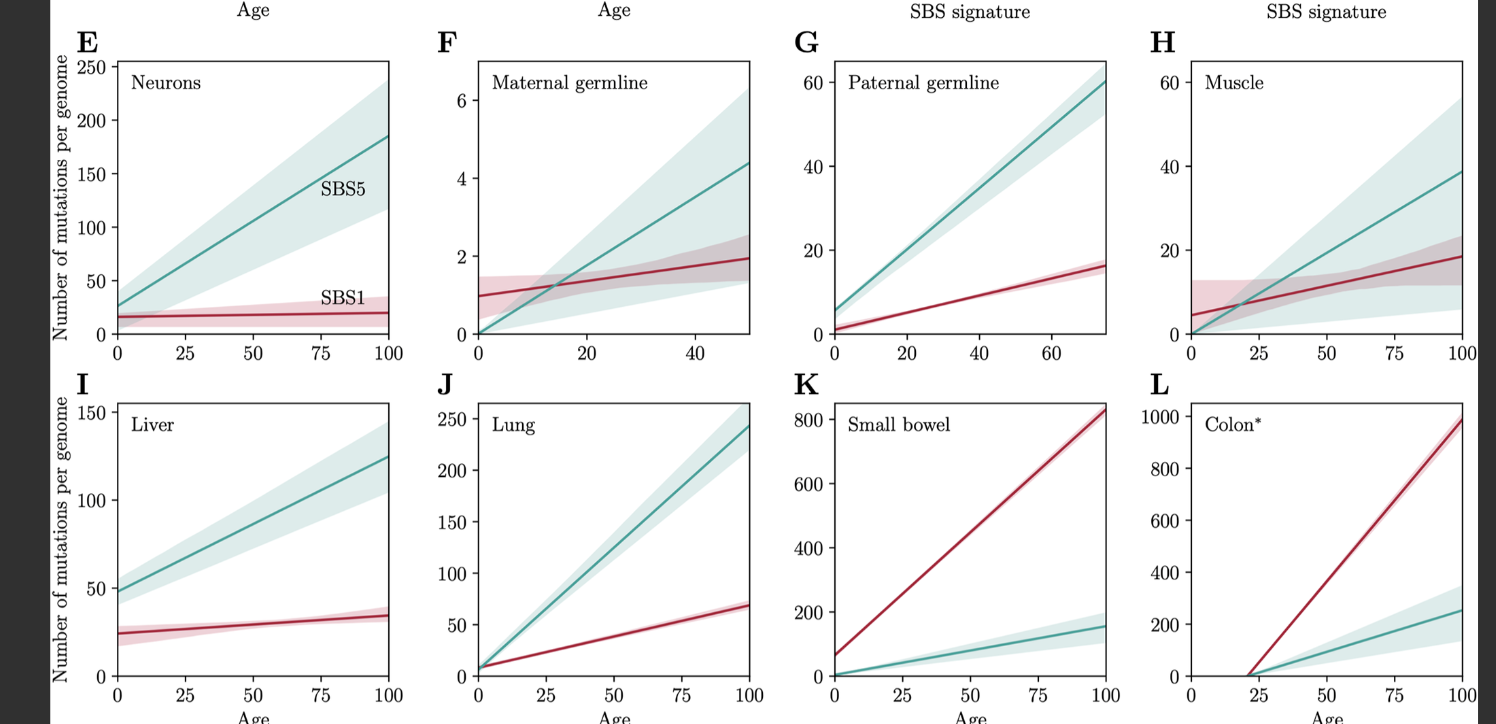
What is this SBS1? It's a signature of mutations from C>T at CpG sites caused by spontaneous deamination. That is, if we have a CpG site, it can either become demethylated, so the CG with the methyl in the C becomes TG. This seems like a good candidate for the "driven by cell division" component of aging, specifically that involving demethylation. Importantly, because it's driven by a mutation, that CpG site is gone and can't be remethylated again! SBS5 might be a proxy for a a cell-replication independent process that aggregates various kinds of damage (Spisak et al. 2025). This rate is also tissue-dependent, so the rate of damage generation is also probably cell-type dependent. The germline as well as muscle seem to age quite slowly. The former is no surprise, the latter is and I wonder why it might be.
We can see here that if we look at absolute magnitude, neurons should also be aging (~225 mutations total vs 300 for lung). Turns out a lot of these mutations are cell-replication independent!
Given what we have seen thus far, if we applied a clock to a region of the brain like the cerebellum that's mostly neurons and see if it ages slower: If a good amount of what the clocks pick up is cell division (through PRC2) as a proxy for chronological age then they may underestimate their real aging.
At a high level, the cerebellum becomes less functional and shrinks in size (Arleo et al. 2024). Epigenetically, the cerebellum does seem to age slower in the usual clocks: It is possible to construct cerebellum-only aging clocks (Wang et al. 2023) and if one applies those clocks to other tissues then one observes that it is the other tissues that age faster. Here's the original Horvath clock (Pan-tissue clock) on cerebellums:
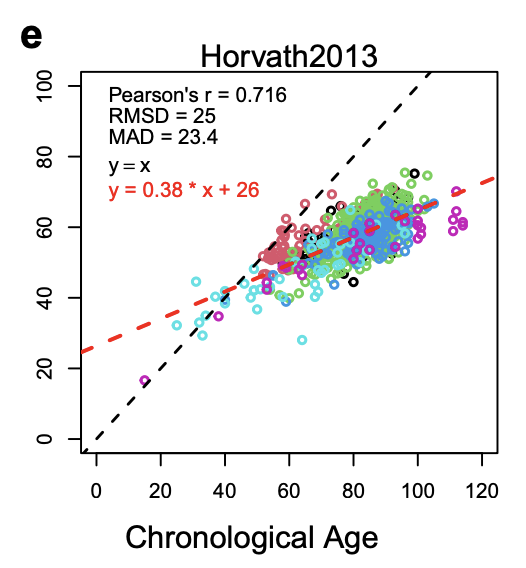
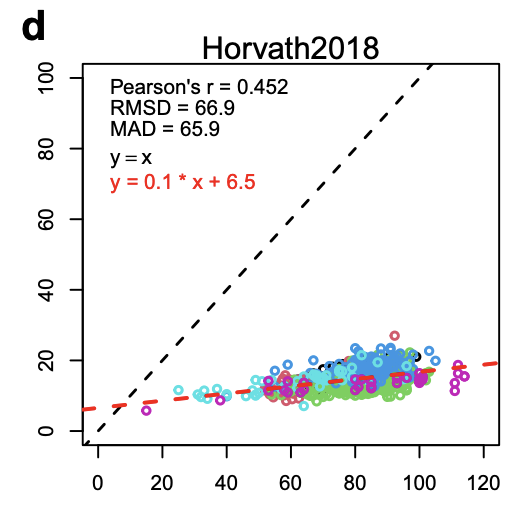
And Horvath 2018 (Skin&Blood clock) is even flatter, perhaps because skin and blood are tissues where there's higher turnover so this clock is picking up more of that cell-division driven aging signal.
When looking at it, they indeed find that compared to other brain tissues, the cerebellum is less methylated at CpG islands, which tend to be PRC2 binding sites, the changes that they do see are smaller and there are fewer that are significant.
One model we could try to explore for neurons is that by and large they don't age and that all variation in cognitive abilities can be explained through neuron loss instead of reduced function: just like with naked mole rats (Ruby et al. 2018), which don't experience an increase in their mortality rate over time, they still eventually die. As we largely lack neuronal stem cells, once a neuron is lost, it is not replaced, which would then explain the decline in cognitive abilities.
But in healthy human beings one doesn't see much cell loss in the hippocampus and enthorrhinal cortex in healthy patients, both areas greatly affected by Alzheimer's in their sample, with rates of neron loss of up to 50% (Price et al., 2001).
Neurons become senescent over time (Hudson et al. 2024) and senescent cells are overall less functional so despite the clocks saying neurons age slower (or not at all), given that cognitive faculties do decline with age, we should take this as evidence that we shouldn't trust the clocks here.
Better is worse: desiderata for a gold standard aging predictor
If you train a model to predict mortality in humans, it is likely the model will learn to pick up correlates of things like diabetes or heart disease. This is indeed what happens with newer generation aging clocks, which I discussed earlier. This would explain why on some of these clocks things like an infection or losing weight reverts epigenetic aging.
An aging predictor ("clock") should take in some data (could be the epigenome, metabolome, etc) about an organism and return a biological aging score. For us to think that the model is measuring cellular aging, the follow would have to be true:
- Chronologically older individuals, on average, present as biologically older by the clock
- The clock ticks in vitro as well, though it may tick at a different speed
- iPSC reprogramming largely resets the clock
- Gold standard age-slowing interventions like caloric restriction or certain knockouts (growth hormone receptor mice) slown down the ticking of the clock
- DNA damage should accelerate the clock while the insult is being applied (eg. radiation, chemotherapy) and progerias should have the same effect
- The score is somewhat predictive of mortality risk and health conditions above and beyond chronological age
- Ideally, but not necessarily, able to do the above across species and tissues
- The model should be able to be trained from in vitro data to predict in vivo data and vice versa
- The model should be examined for causal plausibility; ie it should be known how it works. This need not be causal; eg later I discuss entropy-based clocks where each specific site does not matter, and instead what matter is the higher level noise being measured.
It is acceptable if such a model ends up being a worse predictor of disease and mortality than models directly trained to do this, for reasons already argued.
Somatic mutation accumulation correlates with aging
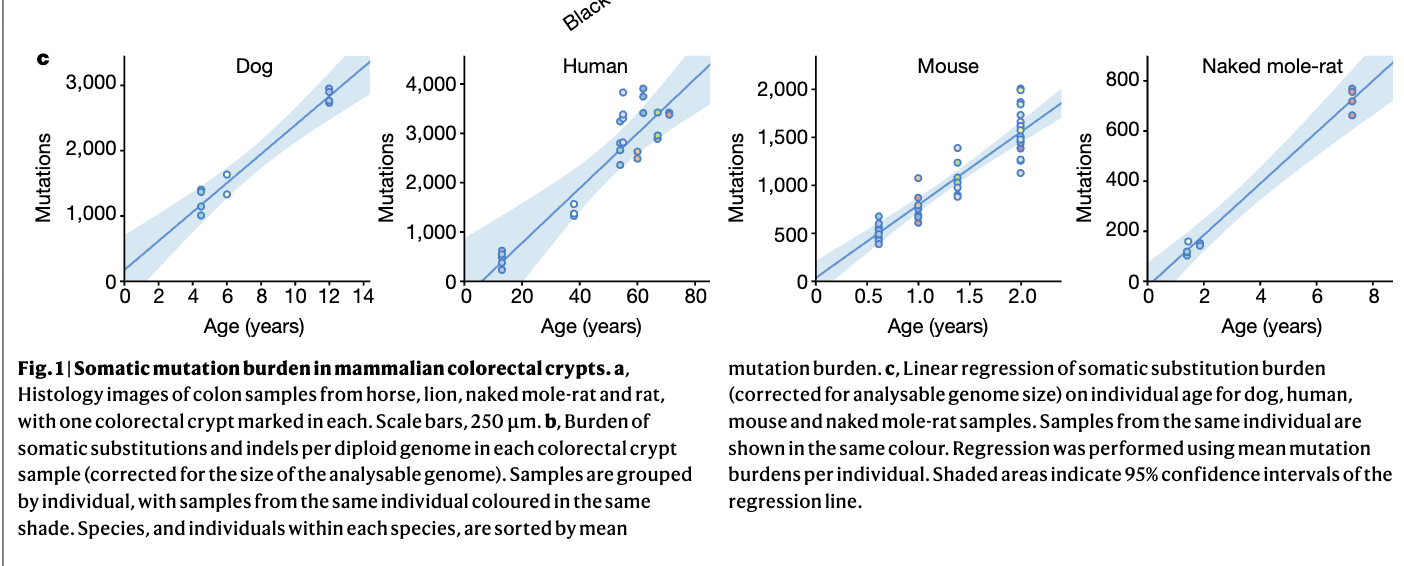
Earlier I said that DNA damage is what drives a good chunk of aging. Worse DNA damage repair also leads to more somatic mutations and their rate of accumulation (single base-pair mutations per genome) correlates with lifespan. This accumulation seems to be linear in a given species.
If one takes those rates for various species and plots their lifespan, one gets a nice curve:
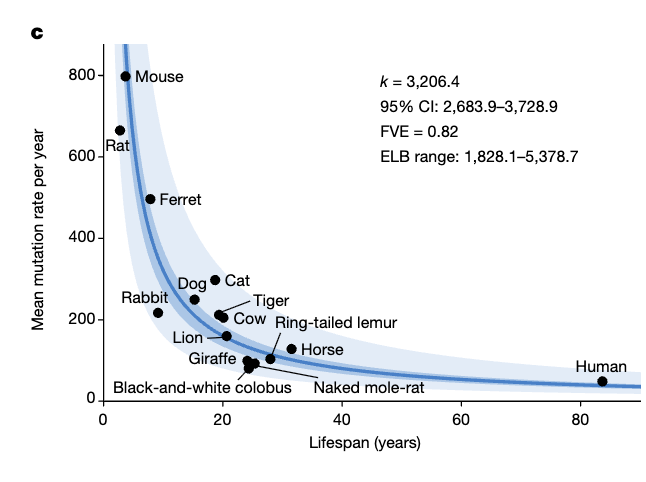
Their equation for lifespan is L(years)=3206/mutations; taking the baseline rate for a given species: Lifespan Fold Change=1/Mutations Fold Change . So for a mouse for example if we were to halve DNA mutations we would get a lifespan of ~8 years, roughly a doubling. The same would be true for humans (half mutations=double the lifespan).
This is for crypts in colons which should be among the tissues that mutates the most (and hence ages faster (and gets more cancer)):
This paper pairs well with Crofts et al. (2023) , a very similar one that does the same thing but with methylation where the result is essentially the same, though they apply it to different tissues (blood and skin) and use a different definition of lifespan (maximum recorded lifespan for an organism). Similar ideas are contained in Bertucci-Richter & Parrott (2023) or Horvath et al. (2024) who also ties this in particular to specific rates of methylation in bivalent (PRC2) promoters (promoter with both repressive marks like H3K27me3) and activating marks (like H3K4me3).
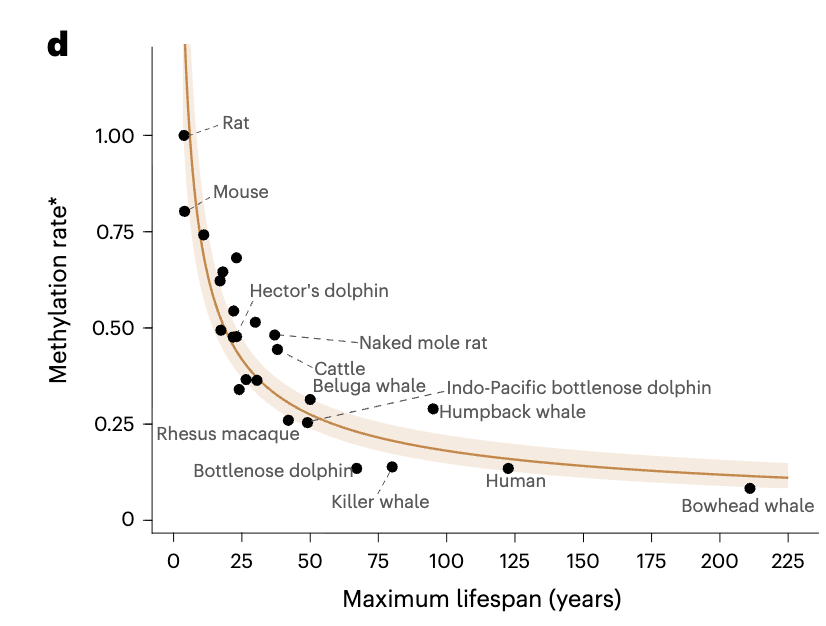
Bowhead whales are quite based entities; as you would by now expect, their cells are remarkably resilient to radiation through better DNA repair (Firsanov et al. 2024). They also run colder which would help explain why it's easier to repair things (less entropy). Their cells divide slower than human's: they take 2x the time to divide and they live about twice as we do. Intriguing isn't it! Does this work in reverse? Do mouse cells take ~40x less time to replicate? Not really! So it's not just just slower cell replication.
Can't we just use somatic mutations (in particular the SBS1 and SBS5 signatures) as a clock? One could try: If you sequence someone's blood cells say, subject them to some intervention for a year and then come back and estimate those signatures, if their DNA damage repair is better, their signature will have ticked up less than controls and from there one could infer slower biological aging. But that wouldn't work for measuring age reversal, as iPSCs retain their mutations but have the rest of the aging phenotype wiped clean.
And are dwarf mice or mice on caloric restriction experiencing less mutations? They indeed are. Are humans undergoing chemotherapy (which also leads to accelerated aging) accumulating more somatic mutations and have a faster ticking clock? Yes to both.
Are cells from longer lived species more resilient to DNA damage? You bet! (Hall et al. 1984)
Somatic mutations and methylation drift are linked because one (the former) causes the latter (Koch et al. 2025). Accordingly, their rates of change vary similarly, and are connected to some extent to cell division, hence DNAme age and most of the somatic mutations occurs during development and then stabilize to a stable rate of ccumulation thereafter (Spisak et al. 2024.)
Axolotls!
Not every living being seems to age similarly from an epigenetic perspective: axolotls don't age epigenetically after development (Haluza et al. 2024):
Here, we conduct DNA methylation profiling of axolotl tissues at CpGs associated with ageing across mammalian and amphibian species. We develop axolotl epigenetic clocks at both panand single tissue levels and uncover that axolotls exhibit conserved epigenetic ageing traits during early life but not thereafter, deviating from the established notion of organismal ageing. We reveal that, in contrast to mammals, the axolotl methylome is remarkably stable and does not exhibit substantial shifts at either global or PRC2-associated gene levels late in life. These findings offer critical insights into the nature of negligible senescence.
Axolotls live to ~20 years (Yun 2021) and don't seem to suffer from much cancer, their telomeres are very long and express telomerase to maintain them (Yu et al. 2022), and a result they don't accumulate senescent cells like we do. Their aging is, as is that of Naked Mole Rats, not Gompertzian but rather their mortality rate is constant through life.
Why do they die, one might wonder! The answer seems to be predation, infections or poisoning or things as silly as choking on gravel. So plausibly, if someone was dedicated to making their axolotls live for 2x or 10x their normal lifespan by having antivirals or antifungals that are safe for axolotls, that might actually be achievable.
Beyond the epigenome: cancer and telomeres
Human beings die and cultured cells get senescent in vitro, but there are cases where cells seemingly just keep growing without ever dying (cells that are made to express telomerase being an example). What does this say about aging?
First, a reason cells stop growing is that their telomeres shorten enough that they enter replicative senescence. Telomere shortening is one of the bottlenecks for continued life that living beings face, but this bottleneck may become a constraint on lifespan before or after epigenetic aging; if it is mostly after then telomeres don't matter much for aging. One way to test this is by giving a cell or an animal artificially long telomeres and seeing what happens. The Blasco lab did that, observing mice live a bit longer, were less diabetic, had lower cholesterol, and were overall healthier. Moreover they also observed that the rate of telomere shortening is related inversely to lifespan, same as DNA damage repair:
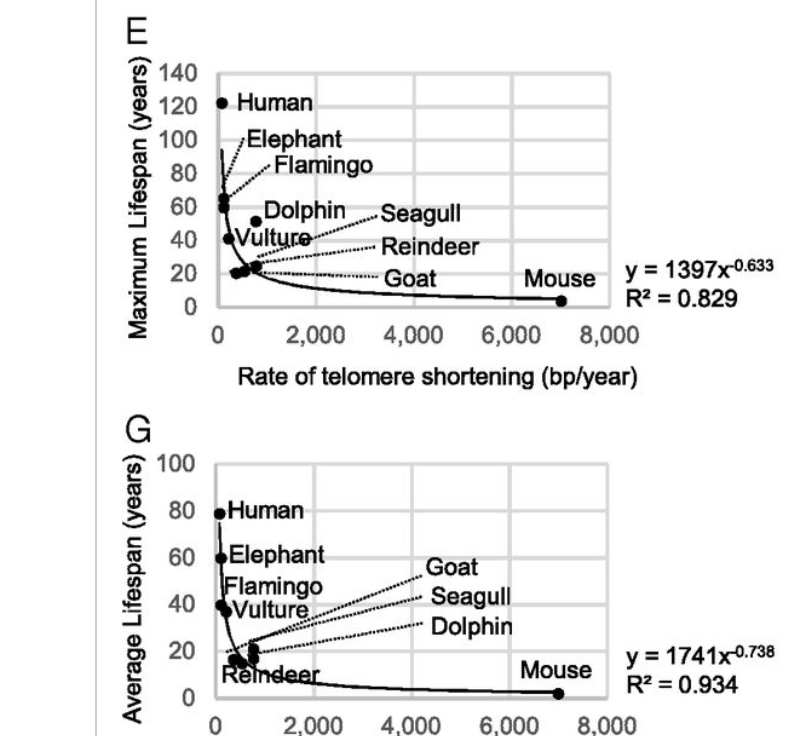
In humans, diseases where telomeres are extremely short exist, Dyskeratosis Congenita being one so we can test what happens if you have very short telomeres. This is useful to see which cells or organs are most affected: predictable it is those with fast turnover like skin or the bone marrow (as opposed to say, the brain where cells don't divide much). These patients can live to the age of 60 whereas patients with defects in DNA damage repair like Cockayne syndrome live to 12, highlighting the relevant importance of these two mechanisms: telomere shortening is bad but not as bad as epigenetic aging as a whole.
Moreover, we know that if one keeps telomeres long artificially, cells continue to epigenetically age (Kabacik et al. 2022) and though full reprogramming to iPSC lenghtens telomeres through TERT reactivation, partial reprogramming doesn't necessarily (Doğa Yücel & Gladyshev, 2024): these two mechanisms can be modulated independently but they are probably co-caused because telomeres shorten not only determinisitcally upon cell division but also with DNA damage, so more efficient DNA damage should also make telomeres shorten a bit slower.
I had said at the beginning that the aged phenotype is an inflammation-like state triggered by DNA damage. Telomere damage as well as shortening is one of those triggers, through the cGAS-STING pathway (Yang et al., 2017, Hewitt et al. 2012, Miller et al. 2025).
Ok so we know we need to eventually fix the telomere issue to progressively increase lifespan, which is tricky to do (Seemingly some compounds can reactivate telomerase so not all hope is list, see Shim et al. 2024). But suppose we just express telomerase to get rid of this problem. What happens to epigenetic aging when cells can just keep going? Do they eventually die of epigenetic aging?
The answer is of course not: If one takes HeLa cancer cells, isolated in 1951, to this day they continue to be used in culture. These cells are chromosomically unusual, having triple the number of chromosomes human cells have. Horvath looked at cancer in his original 2013 paper, finding that cancer cells seem more epigenetically aged per his clock, with ages increasing to 177 years over the donor age in one cell line. These results are more clean with the EpiTOC clock but that may just be cell divisions. I don't necessarily buy that "cancer is more aged".
Plausibly, epigenetic aging breaks down in vitro as cells eventually find ways to survive through mutation and natural selection: epigenetic aging is an issue for us because we want the cells to be in a certain way that fits well in the multicellular whole, but with cancer, cells just have to grow and divide; if a cell fails it dies so as a whole the population becomes truly immortal by constant evolution. Imagine say that some site in the genome. I couldn't find a dataset of cells cultured in vitro for a very long time where epigenetic age is measured but what I expect would happen is that epigenetic aging would continue to increase and then become very noisy: CpGs that used to exist might mutate and disapper, genes or chromosomes might duplicate so CpGs appear twice, etc.
In my What is Aging post, I described aging as a drift away from fitness. Cancer illustrates the limitations of this definitions: cancer itself may be quite fit at replicating but it lowers the fitness of the organism it is in. I think it is still possible to define some notion of aging for cancers but I haven't thought much about it.
iPSC reprogramming as a test for aging measurements
iPSCs are a great construct to study aging. Their epigenetic age is ~zero regardless of the age of the cell of origin, and when differentiating cells out of them, one obtains young cells. If one accepts them as equivalent to embryonic stem cells, it is warranted to assign them a ground truth age of zero, so one could use that to train models to predict age, or to test whether an age predictor yields the correct answer. This may be a unique property of iPSCs, in that if one takes a cell from someone that is 74 years of age, the "real biological age" of their cells won't be exactly 74, that person may have aged faster or slower.
But somatic mutations are still there. Plausibly mitochondria are less functional than that person's original ones because the mtDNA mutations are still there. But other than that, if we could get as rejuvenated as an iPSC gets, that would go beyond anything anyone has ever achieved with any anti-aging intervention.
Not only iPSCs have an age of zero: they don't epigenetically age in culture2
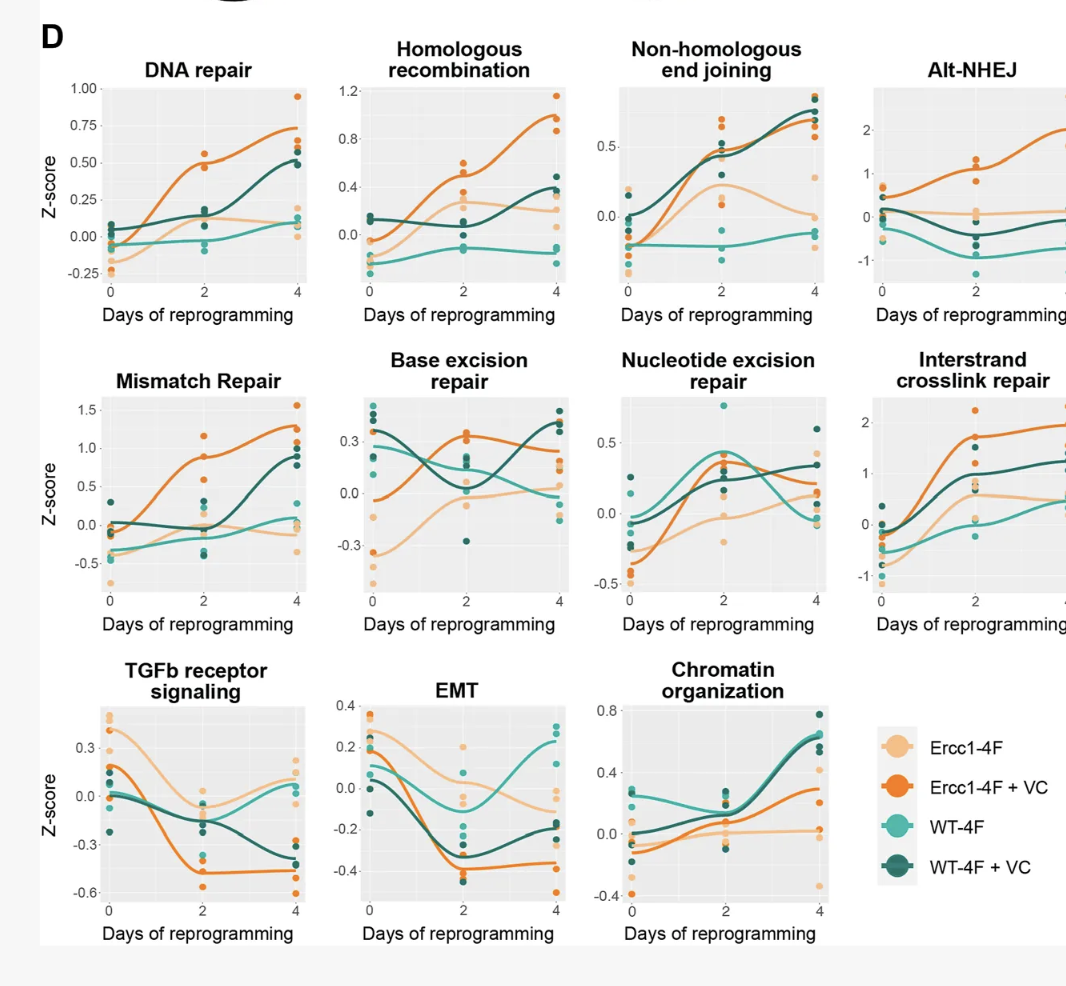
Comparing cellcs in steady as Liedtke et al. 2015 did shows the same results(Here BM MSC=Bone Marrow Mesenchymal Stromal Cell, USSC~neonatal MSC; iPSC=induced pluripotent stem cell). NHEJ, HR, NER, etc are all DNA damage repair pathways.
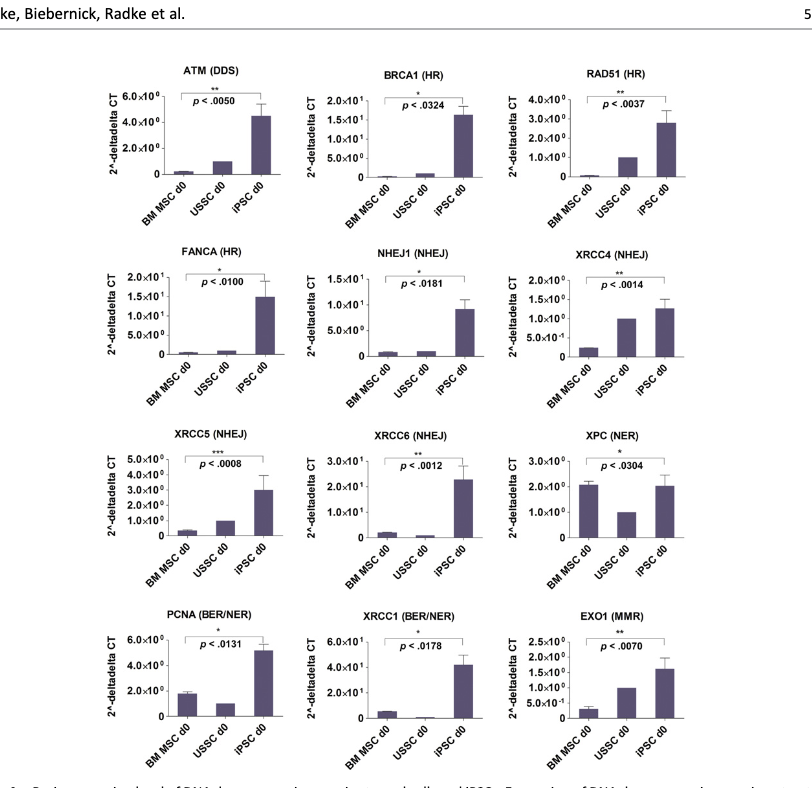
My interpretation of this is that there is capacity already in our DNA to do so much repair that aging can be drastically slowed, if only we find a way to turn that on without also turning a cell into an iPSC. It is another question whether cranking up DNA repair alone would reverse aging, that remains to be tested.
How is reprogramming able to erase epigenetic aging? And what does this tell us about rejuvenation broadly? I used to think that methylation (and the epigenome, broadly) was largely "set and forget" and that the way iPSC reprogramming works is through resetting the epigenome, wiping it clean, and that perhaps the iPSC/embryonic state is uniquely stable or something like that. If that is true, then there would be no way to decouple pluripotency from rejuvenation. Rejuvenation in this case would be resetting, and this is something that cannot be done in vivo. I mean, people have tried, and the outcome is dead mice (Abad 2013).
Of course, people have by now done reprogramming partially, with substantial benefits in progeria mice (Ocampo 2016) and lesser benefits in wildtype mice (Macip 2024) in terms of lifespan. Some benefits have been observed as well for CNS disease models (Anton-Fernandez 2025; Xu et al. 2024; Rodriguez-Matellan et al. 2020). We too have seen some good results at Retro. So it kinda works when done right, but it is limited.
But reprogramming wasn't discovered by searching for rejuvenation, it was discovered by searching for ways to make cells embryonic, which is not what we want.
But what's next? One approach stems from the observation that the TFs that are lost with aging in a given cell type tend to be the TFs that made that cell type such, i.e. "identity TFs". Empirically, re-expressing these TFs rejuvenates cells. For example, HNF4a, if expressed in liver, gets you a younger liver (Yang et al. 2021). This is quite powerful and it already works, but it is limited to specific tissues where one can deliver the relevant cargoes.
Another approach could be chemical rejuvenation. Small molecules have already been used to make iPSCs (Wang et al. 2025) and it turns out to be more complicated than with OSKM. That's good! It means there's more margin of safety before causing cells to de-differentiate in vivo. Some of these chemicals have very recently been applied to rejuvenation. It is still early days, there is only a fistful of papers doing this, but the result establish a crucial "zero to one" proof of concept: it is possible to do things like reducing DNA damage, markers of senescence, and DNAme age purely with chemicals (Schoenfeldt et al. 2025; Paine et al. 2024) though when tested in mice it doesn't quite work yet (Wayne et al. 2025).
Aging: stochastic vs deterministic
Some papers in the clocks literature have pointed that it may be moot to try to analyze this clocks much because some or most of what is observe, they argue, may be due to a random process. You might have an intuition that this is impossible: how could one build clocks then! But that intuition probably comes from assuming gaussian noise with a mean of zero. However, imagine a random walk that is bound by 0 and 1. In that case, no matter where you start, the population average will tend towards 0.5 over time.
And given that we have a trend, one could then train a clock on it, but it would be a clock built purely on noise, epigenetic drift. This model is not true, however as I'll point out in a.second.
So is it just noise? Some might be, but it can't all be noise (in an 'everything becomes mush equally' sense), some of the oldest known age-related CpGs like cg16867657 in ELOVL2 do not look like a random process that tends towards 0.5 (Slieker et al. 2018). Note also that in the cerebellum (mostly neurons, which don't divide) this CpG barely changes, similarly to what's observed with methylation clocks, suggesting that cell division may be be the main contributor to epigenetic aging. The tissues that do age do not show the pattern expected from random drift, rather the opposite is true! Observe that if we only looked at tissues where methylation starts very low (like muscle) then someone could argue that that rising trend is compatible with the stochastic process described earlier, but here we see that for tissues where the methylation starts higher like subcutaneous fat or monocytes, the trends is to continue to increase, not trend down towards a mushy 0.5. Furthermore not only is ELOVL2 like this cross-sectionally but also longitudinally: the trends have a similar slope for many individuals (Figure 4 of Bacali in et al. 2016) And in this dataset containing a bunch of iPSCs, the methylation level for this site (cg16867657) is <10%, as one would expect by continuing to extrapolate to zero.
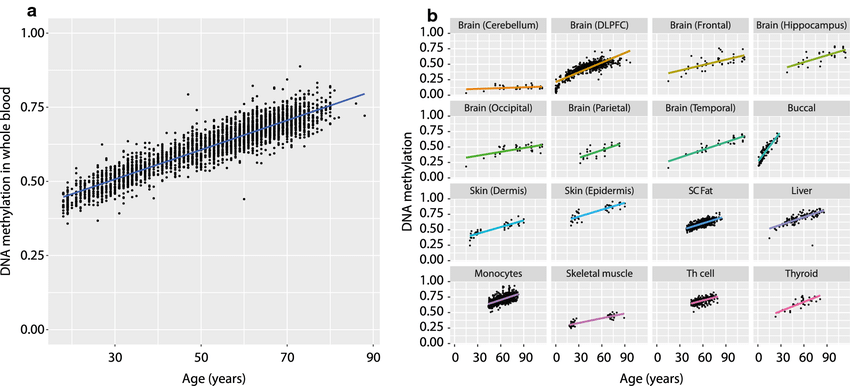
Hence we have existence proof that there are patterns to methylation changes, they are all not random. This is compatible with aging being a process driven ultimately by entropy.
Let's look at papers that posit that what the clocks measure is stochastic change. One thing to bear in mind here is a bit of a political aspect within the field of aging research: there are those with strong opinions about whether aging is programmed (as in it was selected for) and whether aging is stochastic random damage accumulation. Those that say it is programmed take the patterns picked up by the clocks as evidence of a program of some kind, and it seems that as a reaction to that there are some papers arguing otherwise: that it's noise and that noise as I argued above can still lead to a clock. There's then another camp that argues that it's noise and that the regulated patterns we see are damage responses. This was the original view of Horvath in his clock paper.
My own view is: at source it is random, but cells reliable transform that randomness into patterns we can observe at a higher level than individual sites in the genome. Earlier, for example I described the PRC2 sites: The loss of repressive histone marks at those sites is random, and in turn the methylation increase of those sites, though a regulated process meant to replace the histone mark loss, appears random as well even though it's not.
Car aging as a way to understand human aging
To use an analogy, cars are not explicitly designed to break down but they are also not optimized to last forever either. Parts have different durabilities too so on average in the "aging" of a car one will see that the tires and break pads need replacement, then later the motor and then eventually the chassis might rust and fall apart (A similar analogy was used, as I found after writing this paragraph, by Leonard Hayflick in 2007). In some cases some parts might break faster than others by chance or because of a particular environment (imagine someone slamming the brakes all the time, then those might break faster). If one looks at a population of cars one will observe a pattern of parts breaking down in specific ways. However, if one were to look at a part closely (say a brake pad) at some microscopic level maybe that damage is random.
We can draw analogies between car aging and human aging:
- If we were to measure a bunch of attributes of a car (absent part replacement) we would be able to build a clock of how long has the car existed. That would not guarantee us that those attributes are causal. That is, we may be able to infer how dusty the car is from how chipped is the windshielf
- One of those attributes might be the condition of the fake leather in the steering wheel. That may correlate with the "lifespan" of the car but causally, fixing it does not cause the car as a whole to last longer. This is akin to CpG changes are not tied to lifespan but rather some sub-function that is not critical for survival.
- Given replacement however, a clock trained on how worn off tires are would be no longer informative of how durable a car is after one changes them. This is similar to how epigenetic clocks trained on blood would tell someone that their age is that of a child if they were to receive a full bone marrow transplant from a child.
- One can take a composite of metrics of the car (how beaten down it is broadly) and use that to define 'car age'. This would be similar to Pace of Aging or PhenoAge and be subject to the same caveats as those metrics are.
- One could take the same car and sample it over time and do this across a population and notice what tends to predict the rate of decline overall. Perhaps you observe that cars with lots of debris attached to them (markers of the drivers driving them on roads in poor condition) tend to decline more. Then you could build a CarPACE (like DunedinPACE) clock that tells you the rate of aging depending on the amount of debris in the car. The debris wouldn't be driving aging, they would be a correlate of it, and one that's superficial. While starting to drive in better roads would reverse CarPACE, wiping clean your car would do it as well though the rest of the damage would remain.
These examples illustrate that just because some things are correlated in a system doesn't mean they are causally connected (This from Judea Pearl is a good read on the topic), which means that under a causal intervention they may no longer change (the correlation breaks). This happened recently in a trial where before the intervention, epigenetic markers were well correlated with actual metrics but after the intervention they were not (Kou et al. 2025)
And now we can ask: What would a "intrinsic aging" clock in a car look like? The answer to that is the key to understanding "intrinsic aging" in a human. Obviously, the "intrinsic aging" of a car is not a very useful concept. Parts are made of different materials and break at different rates. They are differently affected by the environment. They do share some commonalities: it may make sense to define the age of the tires and all 4 will be related, or the age of the steel of the chassis as a whole, but it makes little sense to bunch those together.
Then, once we have defined the various "ages" we can try to predict outcomes of interest. Not all ages will be relevant for the purpose at hand and in the same way. "Tire age" may mostly not matter for when a car will have a mechanical failure except if they are extremely overinflated and blow up. Perhaps "car lifespan" ends up being driven and predicted purely by "engine age" if that's always the first component that always fail. Whereas "car healthspan" (energy efficiency, aesthetics, other things apart from mechanical failure etc) could be predicted from other "ages" like the aging of the interior plastics, tire age, etc.
Bringing this to humans, paradoxically we hava en easier job because we are made mostly of cells, so if we have cell age we have already captured most of aging. And in turn because cells turn over most of their components, and we know what limits cell function, we can define cell age as a composite score of: telomere length, somatic mutations, and epigenetic state. Out of these we could then try to make predictions, and I expect for many aged phenotypes, it will mostly be the epigenetic age that will carry the most causal power.
Then we can look at other changes with age that we can easily get our hands one: the mechanical properties of our bodies (stiffness, elasticity) are a function of cells and their extracellular environment (the extracellular matrix) which may or may not change with changes in cell age. Of particular interest here could be arterial stiffness as it predicts cardiovascular disease.
Methylation turns over: cars at not cells
Is methylation "set and forget" without much turnover, where once it is lost, it can never come back or is it constantly being adedd and removed?
At a population level, if one measures bulk methylation (as is common when measuring methylation), methylation doesn't change much in the short term (Furukawa et al. 2015) except a small number of sites (Komaki et al. 2021) but that doesn't mean that a given CpG in a specific cell hasn't turned over. As a result, the methylation clocks, if sampled on the same person over and over tend to give relativelt similar results (Komaiki et al. 2022). This paper is also of particular interest to those interested in measuring aging: Eyeballing the charts there means that a change in DNAme aging under 10 years may be noise. This can be overcome if one has a larger sample and one is looking at a population as opposed to an individual; eg in Poganik et al. (2023) they are able to detect changes in DNAme age after various medical procedures; where some of these changes are reversible. I take this paper as evidence that a good chunk of what many of these clocks are measuring correlates of inflammation, and that intraindividual variation can be large and in unexpected directions: Looking at Figure 3 does not inspire much confidence about the clocks!
So if we can't quite track a single cell's methylation level yet, what can one do to see if methylation turns over or not? One can interrogate this question by first looking at the underlying biology: Methylation is known to be established actively by DNMT3 ("de novo") and then maintained by DNMT1. It can be removed by TET enzymes. One can then inhibit these to see what happens. If methylation is constantly being churned, altering the balance of the actiity of these enzymes will result in rapid under or overmethylation whereas if methylation is "set and forget", knocking out those enzymes will matter less. The current understanding is that methylation (Parry et al., 2020 [Reik lab]) at least in stem cells (Ginno et al., 2020), where it has been studied the most indeed does turn over a lot. So a single CpG may be methylated now and lost in 5 minutes, then remethylated later, etc. This is unlike DNA, where a base pair is not constantly flipping back and forth from AT to CG.
As a result, methylation is, as the paper earlier puts it, "not bound by the strict constraints imposed by the rules governing thermodynamic equilibrium". In a closed system, entropy always increases so if there were no enzymes touching the CpGs they would all lose information and turn towards random mush (50% methylation in bulk everywhere) without any hope to recover that. But that is not the case: CpGs are not a closed system. It is this fact that allows for epigenetic rejuvenation we see in reprogramming across the entire epigenome. I want to remark on this again: Cells 'know' to some extent that a given site 'should' be methylated or demethylated, whereas they don't know if a given site in the genome should be an A or a C. Experimentally, one can inhibit methylation and drop it to 50%, then remove the inhibitor and cells can remethylate to some extent back to where they were (Wong et al. 2011). This is again very much unlike what happens if one induces mutations in DNA, they just stay there. As to the how is this happening, the idea of error-correcting codes might give us an idea: multiple histone marks, CpGs nearby, etc can help the overall system keep homeostasis (Bruno et al. 2022; Wang et al. 2020) in the presence of small local disturances. See here for a fun interactive demo.
The reason methylation changes with aging is that cells don't have enough capacity to overcome the drift that occurs. Hence, I predict, if the rate of damage generation were to be reduced, or the turnover increased, it might be possible for cells to put energy back into restoring the epigenome and thus reverse epigenetic age. Some evidence that CpGs with higher turnover are better maintained is Ming et al. 2020, and some evidence that a cell that is hypomethylated (through less active DNMT3) can bounce back when it gets it back is Li et al. (2024).
Hence, whereas both in cars and cells the alterations we observe is ultimately due to entropic reasons, cars have no means to repair the damage that has occurred, they are not constantly remaking themselves whereas we, to an extent, are.
Aging as entropy
At the root of aging there's entropy: The structures holding information (DNA and epigenome) in cells deteriorate over time in a way that is at the same way stochastic and patterned. This is no different from what happens to any system that exists where the rate of generation of entropy is higher than what the system can dissipate.
A consequence of this is that whereas young individuals are very similar in a way, aged individuals (or the aged epigenome) can look very different. In every fetus perhaps a particular site in the epigenome is closed whereas in older people, it will be closed in some and open in others.
Picture 100 glasses of water at room temperature. They all look the same. Now picture 100 glasses of water that are boiling (increasing temperature raises entropy. They all have high level structures (water bubbling) that let you tell that they are hotter, but if you took a picture of them all, the exact place and size distribution of the bubbles would be different. This is what I mean when I say that aging is at its core the product of noise and yet that noise gives rise to observable, predictable patterns across individuals.

Stochastic aging clocks
Let's now look at the papers that discuss the entropic nature of aging.
Mei et al. (2023) from the Conboy lab points out that many CpGs in clocks do not change consistently with age and that when one removes CpGs not picked by the clock from the training set and retrains, then the clock picks different ones. So perhaps clocks might be doing some amount of overfitting? To work around that they posit that certain methylation sites are particularly important if they are invariant with age in general and that if those get disregulated then one would be more aged. They also do something hilarious and quite, dareisay, based: They train a clock to predict the population of the US:
And as we illustrate with the size of US population, non-biological changes can be successfully predicted by the DNAme array datasets. Summarily, any set of regularly changed numerical values can be linearly predicted through EN training on any large data, biological or not, physically connected to the measured axis or not. If anybody is interested, an expansion of the Universe could be predicted from evolutionary DNAme array data. And a person’s health and age could be predicted with relatively good accuracy from expanding distance between the Galaxies, or anything that changes progressively over time.
Since the physical meaning of the training parameters is lost in EN process, any assumption that specific health scores, C-reactive protein levels, etc., are being predicted, is ambiguous, because in the model training, a decline in health, reaching mortality and the increase in age all have linear progressions designated as a regular series of numbers.
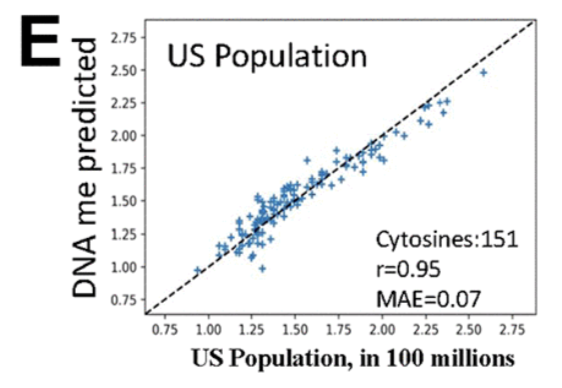
The point being, that just because one can put dots on a line it doesn't mean the effects are real automatically: If you have say 50 samples and you have 450,000 features total, you can fix almost any pattern that is sort of linear. So one has to be careful!
The clocks they construct fit their theoretical model: sites that don't change much with age or sites that have different standard deviation at the population level are all positive correlated with age; ie there is no site that tends to decrease their variance with age in their data.
One disadvantage of this is that one cannot apply this clock to an individual, as it requires a population to calculate the standard deviations for a given CpG, but the authors are not trying to build an aging clock rather they are trying to study the aging process and to show that the clocks others are build are not directly picking up on aging but rather proxies at best.
Then we have 3 papers, all published May 9th 2024 from Meyer & Schumacher (2024) , Tong et al. (2024), and Tarkhov et al. (2024) [who's now a coworker at Retro!]
I'll take two of these examples: Tong et al. set out to see how much of the accuracy in known epigenetic clocks like Horvath's is driven by aging as a stochastic process vs other causes, finding that Horvath's clock is fairly well attuned to this stochasticity (66-75%) whereas PhenoAge is less so (63%) which makes sense as PhenoAge was calibrated to predict health and disease. Though they don't look at GrimAge or DunedinPACE in this paper, I expect that those would be picking up even less the entropic component of aging and more the downstream diseases. They note that the way methylation changes with age is not orderless but rather there are patterns in that damage (CpGs that are marked by the Polycomb Repressive Complex getting hypermethylated being one example).
Their methodology is quite neat, they pick the CpGs for a clock, they run a stochastic process (a random walk informed by the average change of those CpGs), sample a bunch and train a clock on those stochastic changes, then do the ratio of the r^2s of both clocks to see what % of 'predictive power' is driven by the stochastic process. One has to note here that that doesn't mean that "75% of aging is stochastic", that would be assuming that the Horvath clock is a perfect aging predictor, rather the fair claim to derive here is that a lot of the signal behind the age-related CpGs that the clocks pick up can be modeled as a random process. One objection to this methodology though is that the Horvath clock was trained on many tissues and cell types. In my view aging has a strong cell-type specific component and so any such signal would not be picked up in Horvath's clock or these stochastic clocks.
Something worth noting (that the authors acknowledge in their conclusion) is that these clocks seem to break in unusual cases: Whereas Horvath's clock predicts well age regardless of tissue or age, and predicts age=0 for iPSCs, the stochastic clocks predict these cells to be much older than they are, suggesting perhaps that the stochastic component of aging becomes more predominant during or after development.
But interestingly, whereas PhenoAge is lower in smokers or COVID-havers, the stochastic clocks is impervious to these perturbations. And that's good! It means they are less attuned to transient, easy to reverse changes (perhaps just inflammation) and properly calibrated to the underlying entropic component.
They also analyze a clock trained on in vitro data, EpiTOC2 which should be by construction more free of cell composition effects or organismal diseases, and find that this clock is almost completely driven by the stochastic component of aging.
When one looks at Horvath's CpGs, 160 get hypomethylated with age. Those one could attempt to explain their loss via a passive process. But the other 193 are actively getting methylated; and those tend to be around PRC2, the same CpGs heavily enriched in EpiTOC2. As I mentioned earlier, though which CpG might change is random, the overall pattern once again is not: over and over we see this trend of PRC2 sites getting methylated.
Tarkhov et al. have access to scDNAm datasets (of muscle stem cells) which is a rarity! That way they are able to look at populations of cells at the same timepoint and see how distinct or the same they are and from this to be able to better look at aging. Here they define a metric of coregulation for CpGs, do CpGs tend to change at the same time or do they change independent of each other? In a stochastic model where every CpG drifts individually one would expect little coregulation whereas in development, where methylation is being set in a very opinionated way by developmental programs one should expect high coregulation. This is indeed what is seen and when applied to aging, 92% of the 502 CpG in their dataset that changes with age changed per the stochastic model.
These changes are not uniform in the genome though, the paper notes that the areas that tend to lose methylation are nonfunctional regions (plausibly, transposable elements, another regular pattern) or introns whereas the areas that gain methylation are CpG islands, coding sequences, exons (broadly useful regions).
As a final note, I came across this paper from Vershinina et al. (2021) that claims that 90% of the CpGs are deterministically changing, the opposite conclusion as in this papers, trying to make a case for a more programmed (ie. development-like) pattern to aging. However, I don't think they can really draw this inference from their bulk DNAme datasets of twins which would show averages of populations, hiding away the noise, whereas Tarkhov et al., with single cell datasets can actually look at the issue with deeper resolution.
If this point is not clear, I made this small app where you can visualize an example of this where the ensemble average looks very smooth, almost deterministic, yet the process can be quite random when looked at a single cell level.
One objection to this analysis is that it's done at the methylation, not chromatin accesibility level, and this analysis could overturn the conclusion: Imagine, for the sake of the argument that we engineer a cell to deterministically express an enzyme that will methylate the promoter of the gene COL1A1. The way this will happen is the enzyme will recognize the COL1A1 site and, stochastically, deposit some methylation there. The specific site may be random under some analysis but the overall intent of the program (making the site less accessible) will be fulfilled.
Now imagine you are a cell and you are trying to switch off collagen production. Do you do this by setting methylation in a specific way or do you make a bit more of the relevant enzyme? The end result is deterministic: less collagen but how to get there is stochastic. Out of chaos, order. This ties to the ideas elsewhere in this essay about error correction, where multiple states can correspond to the same useful phenotype. Here in Borgel et al. (2010) where they look at development, it illustrates this idea where specific promoters that are relevant for development are overall more methylated but not deterministically so. Everything in biology is ultimately stochastic and directedness is not a binary but rather a matter of degree. Developmental programs are fairly strong and aging drift or response to aging can be more weakly controlled.
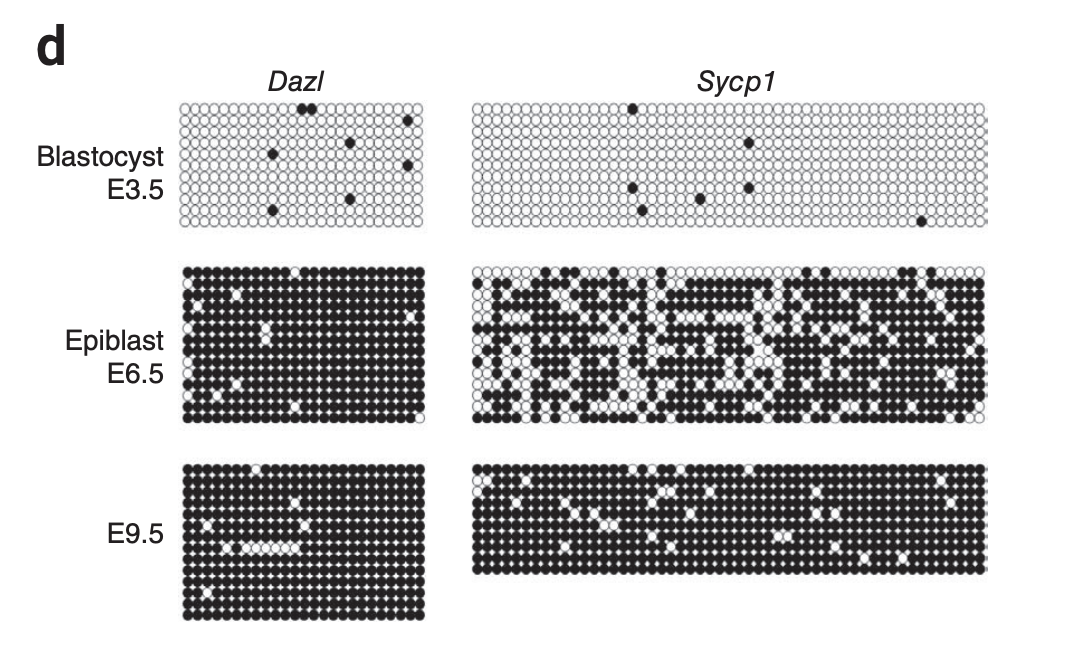
To overcome this issue we could look at chromatin. Unlike methylation, chromatin aggregates all of the epigenome, and chromatin accessibility better correlates with what ultimately matters: gene transcription and protein translation. However, this essay is already quite long so I'll have to skip that here! I'll just have one pointer: Even though the cell is suffering entropic damage, the ways in which cells deal with this are surprisingly similar: PRC2 hypermethylation, retrotransposon derepression, and hypomethylation of useful genes. Aged cells of a given cell type are dysfunctional in different ways at the micro-level (which CpG is (de)methylated) but similar at a macro-level. For more I refer you to Patrick et al. (2024).
The epigenetic magic wand: a thought experiment
Suppose that you believe that reversing the chromatin state of a cell to youthfulness will rejuvenate a cell if not completely, completely enough for most practical purposes. And suppose you have a magic wand that does just that to all cells. Would that restore a person to what they were years back? Would, in short, reverting cellular aging also reverse organismal aging?
I believe this is closer to true in C. elegans (who have a fixed number of cells, no cancer, no cardiovascular system, no stem cells) and less true in human beings. But it is still very close! One can try to enumerate what might not get fixed post-epigenetic magic wand and there are things:
- First, some cell populations are just lost so there isn't much to rejuvenate. This is nontrivial:
- Loss of neurons (Parkinson's, Alzheimer's)
- Loss of sensory receptors (Like hair cells in hearing loss)
- Loss of eggs
- Then there's high level tissue restructuring:
- Fibrosis (lungs, liver)
- Loss of high level structures like teeth
- Crosslinking/stiffening of tissues (Elastin does not seem to turn over much)
- Nephron loss in the kidney
- Spinal disk degeneration
- Calcification of arteries and heart valves, accumulation of plaques
- Thymic involution
- Lastly, there's somatic mutations that still wouldn't go away.
Some of these problems are easier to solve than others. Without attempting to be comprehensive:
- For small cell populations one can in a targeted way replace them out of iPSC. This is already in the clinic for Parkinson's as well as Type 1 Diabetes. There are companies working on eggs. And some has been written about using transdifferentiation to make lost cell types out of existing cells.
- For fibrosis, as per Uri Alon's work and given it does turn over, I am not too concerned that we can find ways to modulate the process to get rid of it. One recent win here is Resmetiron for liver fibrosis, reversing something that merely losing weight with GLP1 does not.
- For plaques, though statins help massively if one starts early enough, there are some attempts at engineering macrophages
Cancer would still remain as a sort of final boss for this battle where I personally have the greatest uncertainty. The easiest approach would be to replace tissues with new iPSC-derived tissues, ideally from banked cells from the patient that have a lower burden of mutations. That itself sounds hard! But even harder it developing a cancer treatment that deals with cancer in situ systemically and reliably. But I have spent way less time thinking about cancer than I have about the rest of aging.
That is, it is possible that after the epigenetic magic wand has been waved someone would have the functional capacities of a 20 year old yet still die of cancer at the same rate as usual, that is: it is conceivable to restore healthspan without affecting lifespan if the epigenetic reset does not do anything for cancer. If, however, we had effective cancer treatments on top, then we would see more meaningful lifespan extension.
The intrinsic randomness in lifespan
When one looks at a large human population, one observes a range of lifespans. Some people live only a few years of age, others beyond 100. That environmental factors play a role in aging is unquestionable, but their existence raises the question: in perfectly ideal conditions where genotypes are controlled and the environment is likewise controlled, how stochastic in aging? We can look at this in two model systems: worms and inbred lab mice. As you can see, the variability is substantial even after controlling for these factors.
What we do know is that the mortality rate increases predictably at a population level and from there we can infer how "loaded the mortality dice are", it's still a roll of dice. Someone who is 10 years old is >99% likely to make it to over 80 whereas someone who is 100 it's more of a coinflip (50%).
So prima facie, even with the world's best epigenetic predictor that tells you someone's legit biological age, it would be far from being perfectly accurate. But that's fine for many purposes. If we are measuring aging interventions and our model tells us that someone risk of death is 50% and we do something and now it says it is 1%, that would be very useful to have, even if does a poor job at predicting when exactly a specific person will die.
Aging interventions that slow down cellular aging are mostly DNA damage reducing interventions
Some age-slowing interventions target things like senescent cells and rejuvenate an organism by depleting less fit cells, but not by rejuvenating them. Others rejuvenate organisms by adding back broken components (stem cells, mitochondrial transplants). But the ones that matter for cellular aging all work on the same principles:
Looking at worms one can see more cleanly what affects cellular aging. Largely, it is mostly damage repair of DNA (as well as autophagy, secondarily) that interventions seem to converge on.
- Caloric restriction (And the whole IGF1/mTOR paradigm) ultimately converges on the upregulation of autophagy (via TFEB/hlh30, downstream of mTOR) and DNA-damage repair (via FOXO3/daf16). As a result, CR slows down epigenetic drift and thus aging (Maegawa et al. 2017)
- As far as people have looked, this effect is small in humans (Scholler-Mann et al. 2020; Waziry et al. (2022)), therefore the effects of the modulating the IGF1 signalling pathway are unlikely to be a promising way to increase lifespan in humans.
- Rapamycin is more unclear. The effects on healthspan don't seem to be there (Horvath et al. 2021, Minton et al. 2024) though a recent conference talk said it extended lifespan 15% in marmosets by 15% (Rapamycin news). There may be an effect size, but a small one, and one that can't be scaled.
- Long lived worm or mouse mutants have loss of function mutations in nutrient sensing pathways, switching cells into repair mode, which leads to the observed effect.
- Interventions that reduce body temperature extend lifespan through increasing accuracy of biological processes, including DNA damage repair.
Other interventions like heterochronic parabiosis are in a way a cheat code to lend extra organismal capacity to an older organism and as such one can say that they can transiently rejuvenate non-cellular aspects of an organism (its blood) without rejuvenating its cells. From the discussion on turnover rates above, it follows that without affecting DNA or chromatin, changes that work through proteins are bound to be short lived instead of permanent. This is indeed what's observed empirically (Poganik et al. 2023).
This knowledge can be and is being leveraged to tackle aging (Tomusiak 2024). Any other approach aiming at slowing down cellular aging will probably not be successful (with the posisible exceptions of mitochondrial replacement and telomerase-based interventions in applications).
Appendix: Proteomic clocks
Methylation is not the only data modality one can use. To me it seems good enough to pick up cell aging, as the methylation is methylation of those cells, and everything else (predicting brain from blood) is a correlation.
One can also measure the levels of certain proteins in blood and use that to infer aging of an organ. The use of proteins as biomarkers is something that is well established in many contexts: ApoB is an even better marker than LDL cholesterol for heart disease, Neurofilament Light Chain is a general marker for CNS diseases, troponin for heart injury, ALT/AST for liver, CystatinC+Creatinine for kidney, etc. These proteins tend come from specific tissues so they are a much better direct readout of tissue health.
One paper (Argentieri et al. 2024) did this, getting results on par with the methylation clocks, but I argue this paper didn't really utilize proteomics to their fullest: they did not try to go organ by organ but rather they built a model to predict chronological age.
This one (2023) from Wyss-Coray is more interesting. Here they first look at gene expression in different organs to tease out which proteins are most likely to be tissue-specific. Then one can use those subsets to train various models to predict chronological age in a sample of ~5.6k people. So for examaple one wouldn't use Cystatin C in a model of brain aging, even when they are correlated (because aging makes brain and kidney aging correlate), because it is not unique to that organ. In the paper they go beyond using chronological age as an endpoint in one case, training a proteomic clock using brain-specific proteins to predict a behavioral cognitive endpoint. This model turns out to be better than age, polygenic risk scores, and pTau181, a direct marker of AD. If curious about what these proteins they are picking up are per organ, here they are.
This other similar paper from the Gladyshev lab (2024), using the larger 53k UK Biobank dataset, and following a similar approach of looking at proteins that are enriched in particular tissue. Needless to say, they are able to train reasonably good models for chronological age and mortality, and these models turn out to much much better than GrimAge; by extension they are probably better than other methylation clocks as well. Oh et all is the Wyss-Coray lab paper from earlier.
On top of that they find that each organ's clock is a better predictor of disease in thar organ:
After that, in this preprint, also from Gladyshev, they build proteomic clocks that try to predict disease (instead of chronological aging) and they use neural networks instead of linear models though that was only outperformed linear models for brain aging. This all done with just 21 proteins, which are easier to make into a test people could use. To my surprise, they are also able to predict tissue-specific cancer! In fact they are able to do so using single proteins, no complex models required. Here, just ENPP7 or CDHR2 (adjusted for age) seem to do a decent job compared to the earlier model (the GTEx 4x from the previous paper)
They are better than models built out of simply blood chemistry in many cases but not cancer.
Cancer may be hard to predict because we don't really have interventions that have been deployed at scale that slow down DNA damage accumulation. But even if we had them, it's unclear how those would be reflected in the proteome.
Summary
Based on the above, here are some concrete points:
- Biological aging clocks and personal use
- Commonly available methylation clocks do track to some extent health and mortality and thus a reduction in those scores, all else equal, would be a good thing.
- However, if one's interested in tracking one's health, I advocate for the standard direct measures of clinically validated biomarkers. I would go further and say that these markers and a few others (thyroid function, vitamin deficiency) are better because they are more actionable. Would you rather know that your epigenetic age is 1 year older or that you are vitamin D deficient? For me clearly the latter, as it points to a way to fix that.
- Proteomic clocks, once adopted at scale, will be able to enhance this baseline
- What methylation clocks measure
- These clocks measure different things depending on how they were trained; as a rule of thumb clocks trained to excel at predicting mortality and health in humans will be biased towards disease, not cellular aging.
- It is misleading to compare "how good a clock is" by how well they predict mortality or disease. "How good a clock is" requires a "for what".
- A lot of what clocks end up measuring is PRC2 methylation which is an adaptive response and therefore not causal. However, that can still be useful to measure the rate of aging. I'm less sure whether one should use these to measure rejuvenation, as one could achieve functional rejuvenation in every sense while keeping the age-related PRC2 sites in place. OSKM happens to wipe this out, but that's not guaranteed of every age-reverting intervention.
- For researchers
- Methylation clocks are a valid way to study aging. I would recommend using a panel of handpicked CpG clocks (Like EpiTOC/PRC2-AgeIndex or the Retrotransposon clocks) over pure regression models like the Horvath clock for better interpretability.
- iPSCs/ESCs, and reprogramming are a great way to validate clocks. A chronological aging clock should assign these an age of 0
- It is useful to build cell type-specific clocks for in vitro research that consider how that cell type ages; eg for fibroblasts one can have a collagen genes clock to measure whether that is rejuvenated.
Citation
In academic work, please cite this essay as:
Ricón, José Luis, “From chaos, order: On the nature and measurement of biological aging”, Nintil (2025-09-27), available at https://nintil.com/measurement-of-aging/.
Comments