An introduction to Alzheimer's Disease
Table of Contents
- Introduction: NIA-AA 2018's Alzheimer's Disease
- How did this definition came to be? A history of Alzheimer's research under the lens of the ACH
- Measuring brain function
- What is Aβ?
- What is tau?
- The topography of Alzheimer's
- Amyloid and tau co-causing Alzheimer's
- (A->T)->N?
- Conclusion
- Appendix: An alternative way to produce Aβ without BACE1?
- Acknowledgements
- Changelog
This post, and some that will follow are my attempt to reconcile the repeated failures of Alzheimer's drugs in the clinic with the evidence that the general consensus in the field that the amyloid cascade hypothesis (ACH) is true. Ultimately, what is really causing Alzheimer's, and how might it be stopped? This post will not critically discuss evidence for or against the ACH, rather it will explain what the current accepted definition is, and how it came to be. I know everyone wants to read an analysis of clinical trials and see exactly what aducanumab did or did not do, but that will have to wait for the next post! Theree is a lot of background material that is needed to put those trials in their right context.
Introduction: NIA-AA 2018's Alzheimer's Disease
The canonical definition of Alzheimer's Disease, as used in the field, is inextricably linked to the ACH, to the point that the concept of studying Alzheimer's without regard to amyloid beta is seen as a contradiction in terms. One researcher, Karl Herrup that is critical of the ACH reccounts that back in 1992 he found himself in a situation where
Finally, the chair of the committee spoke, and I still distinctly remember that I was given a strict warning. “Son, if you’re not studying amyloid, you’re not study- ing Alzheimer’s.”
Today, if someone says Alzheimer's Disease (AD) in the academic literature they almost surely mean AD as defined by the National Institute of Aging (NIA)-Alzheimer's Association (AA) 2018 research framework (Jack et al., 2018). This document defines AD and related terms like preclinical AD, dementia and Mild Cognitive Impairment (MCI). The first important point this document makes is that AD is to be defined by a series of neuropathologic changes, to be measured using molecular biomarkers in vivo, and not by clinical symptoms.
The reason for this is that the set of syndromes that we think of when we think of AD like amnesia could have many causes other than AD. Additionally, one could have an early stage of a disease (Where there are no symptoms), but where one can recognize signs of the disease and forecast that eventually clinical symptoms will apear. Think of cancer: Cancer is not defined by subjective symptoms a patient reports like fatigue or having a lump under the skin, it is defined by the presence of a sufficiently large number of cells presenting an oncogenic phenotype.
These biomarkers are grouped in the AT(N) framework, for Amyloid, Tau, and (N)eurodegeneration/neuronal injury. A later section reviews these biomarkers:
- Aggregated amyloid, to be measured by Cerebrospinal fluid (CSF) Amyloid Beta Peptide 1-42 (Aβ42) or ratio of Aβ42 to Aβ40. As well as Amyloid PET.
- Aggregated tau (neurofibrillary tangles, NFTs), to be measured by CSF phosphorylated tau, and Tau PET
- Neurodegeneration, to be measured by anatomic MRI (brain volume), FDG PET (measuring glucose consumption, more dead neurons=less consumption), and CSF total tau (This also goes up in all sorts of diseases that cause neuronal death. There's tau inside the neurons, bound to microtubules, and dead neurons plausibly leak out tau when they die)
These are measured because the ACH is built on these three building blocks: First, Aβ aggregates appear. These by themselves do not lead to neurodegeneration; rather they increase aggregation of tau into NFTs, and it is these that end up directly causing neuronal death. It is this neuronal death that then causes the clinical symptoms like amnesia.
On the AT(N) framework, there are various combinations of biomarkers one could present:

Note that A+T+ (That is, presence of amyloid and tau), regardless of neurodegeneration is classified as Alzheimer's. Tau with neurodegeneration but without amyloid would be classified as something else: The tauopathy needs to be triggered by amyloid for the system to classify it as Alzheimer's.
When one adds cognitive functioning, the classification insists on separating these from Alzheimer's: So someone who has trouble functioning independently has Alzheimer's with dementia whereas someone that is cognitively unimpaired (CU) but has amyloid and tau would have preclinical Alzheimer's. Someone with just amyloid and alzheimer's, but no tau pathology is dubbed Alzheimer's pathologic change, with dementia.
Though formally inspired by the ACH, the framework does not assert that A->T->(N), but it hints at it.
What the framework does do is to tie the name Alzheimer's Disease to the presence of amyloid aggregates, and tau fibrils. No A+/T+, no Alzheimer's. The NIA-AA 2018 guidelines are the results of decades of thinking about the disease and express the prevailing opinion of the field, explaining why Herrup was told back then that if one is not looking at amyloid, one's not looking at Alzheimer's.
My own view on definitions
Arguing too much about definitions is a sign of a diseased field. Definitions are meant to be helpful handles in specific contexts for specific purposes, but invariably naming things has power, which is why debates over what equality, justice, or fairness means will never end. More debate over naming means more politics involved. The ultimate guiding end here that we all can agree on is helping people live better lives, by means of improving or slowing down the worsening of cognitive function, in turn by means of removing cytotoxic substances from the brain. There are many ways to improve cognitive function, and there are many substances we wish to be able to remove from the brain. Specifically pointing to prevention of worsening of function by removal of substances, and pointing to specific substances seems something worth of its own name.
This is compatible with other causes leading to neurodegeneration, or other substances we may want to remove from the brain, or another factor causing both amyloid and tau pathology.
As definitions play a role in coordinating, it would not be in our best interests to propose an alternative definition, as that would lead to communication issues with everyone else. In what follows I will use the terms AD, MCI, CU, and so on in accordance with the NIA-AA 2018 framework.
An alternative way of defining AD is to stress the clinical symptoms. Karl Herrup prefers to define it thus:
Alzheimer’s disease is a late-life disease that, over the course of many years, destroys normal brain function in a progressive and irreversible fashion. Throughout the advance of the disease, the person is largely unaware of the dramatic nature of the changes that are happening to him or her. The inabil- ity to form new memories is one of the first hints that there is a problem. The person then begins to lose his or her ability to perform complex tasks. As the disease progresses, language skills and reasoning deteriorate as does the abil- ity to make judgments. Personality changes such as depression and apathy set in along with unexpected emotional outbursts, aggression, and agitation. The ability to navigate worsens, leading to helpless wandering. For each of these changes, the severity of the dysfunction increases with time. Through most of the disease process the physical health of the person with Alzheimer’s remains strong despite the continuing mental deterioration. By the end stages, however, the person becomes bedridden, incontinent, nonverbal, and nonresponsive.
However, as the NIA-AA report points out, the issue with this is there can be many biological reasons why one may present those symptoms. One needs to go deeper than what a clinician can see externally. Only by looking at the underlying biology can one make progress in treating the disease.
How did this definition came to be? A history of Alzheimer's research under the lens of the ACH
Herrup's book has a narrative account of how we got to this. I enrich this narrative with Hardy's A Hundred Years of Alzheimer's Disease Research (2006):
In the beginning of time (1906), clinician and neuroanatomist Alois Alzheimer identified "plaques and fiber-looking structures" in a patient's brain that presented dementia. It was unclear what those were but soon after, professor Emil Kraepelin included this case in his recently published book Psychiatrie and named it Alzheimer's disease. Admittedly, it was perhaps a bit premature to christen a new disease based on a single case study. Back then nothing was known about what exactly the plaques were, or the genetics or the disease or anything else.
Over time, more cases were collected and it was noticed that some families tended to present more cases of Alzheimer's, so a distinction was then made: Familial AD (fAD) as opposed to sporadic AD (sAD). Eventually, with the advent of progressively better tools and molecular biology itself, the search for potential genes and the identity of the molecules involved in the disease started.
Hardy (2006) adds that at some point scientists found out that many cases of senile dementia presented precisely the same pathological changes discussed by Alois Alzheimer's:
The first autosomal dominant cases of Alzheimer's disease were described by Schottky, 1932, Van Bogaert and Maert, 1940, and Essen-Moller, 1946. Although the family described by Schottky was lost to followup, the families described by van Bogaert were used over 50 years later in chromosome 14 linkage studies (Van Broeckhoven et al., 1992), and the family described by Essen-Moller underwent one of the first documented instances of genetic counseling when their disease was found to be caused by a presenilin mutation (Gustafson et al., 1998).
The modern era of Alzheimer's research started with the realization that the majority of cases with senile dementia actually had Alzheimer's pathology, changing Alzheimer's disease from a rare neurological curiosity to a major research priority. In a series of influential papers, Blessed, Tomlinson, and Roth made the observation that the majority of cases of what had been called “senile dementia” had the plaque and tangle pathology of Alzheimer's disease (Blessed et al., 1968, Tomlinson et al., 1968, Tomlinson et al., 1970). While the separation between “Alzheimer's disease” (with an age of onset of <65 years) and Senile Dementia of the Alzheimer Type (SDAT) (with an age of onset of >65 years) was maintained for several years, it became clear that understanding the biology of Alzheimer's disease would help in the development of an understanding of the causes of dementia in the elderly, and the nosological separation was dropped (Katzman, 1976). The formation of Alzheimer's Associations in the United States in 1979 and later in other countries and increased research funding followed from this realization (Khachaturian, 2006).
On the gene side, scientists found a series of genes associated with the disease. The most important one in sAD is APOE, a gene coding for a protein involved in shuttling things between cells, including lipids and Aβ itself. A specific variant of APOE (APOE4) leads to substantially increased odds of Alzheimer's whereas APOE2 and APOE3 being protective. There are a few other genes associated with sAD but they don't raise the odds of getting the disease by much. APOE4 increases the odds of AD by ~3.3x whereas the next SNP of relevance, rs75932628 in TREM2 increases it by ~2-2.5x (Kunkle et al., 2019). TREM2 is a less well known gene expressed in glial cells, and one of the targets being pursued clinically besides amyloid generation (Schlepckow et al., 2020).
George Glenner and Caine Wong (UCSD) isolated Aβ and published the sequence for 24 of the amino acids in Aβ in 1984. Around the same time it was noticed that Down Syndrome patients also had the exact same amyloid in their brain (With just 1 amino acid being different). Two years later, some labs had found the sequence of DNA that could produce that peptide, which was in chromosome 21... the same one that was known to be duplicated in Down syndrome. That made sense: Double the chromosomes, double the amyloid, faster progression of the disease. The protein was eventually found (Masters et al., 1985) and dubbed Amyloid Precursor Protein (APP), with 695 amino acids of length. Today, we know more about how DNA becomes proteins, and the fact that the same length of DNA can give rise to transcripts (mRNA) of different length. The longest APP isoform is known to have 770 amino acids. The Alzforum website has a helpful diagram of this latter longer protein and the effects of various mutations along its length. The one originally identified is now termed the "c isoform". It was hypothesised that this protein, because of its structure (To some extent you can fold proteins in your head), would be sitting at the cell membrane.
Then in the 90s the genes involved in fAD were identified: all families with early onset Alzheimer's had mutations on the same genes: Always APP, PSEN1, and PSEN2. Not BACE1! This implies that there is already plenty of β-secretase to cut all the available APP, and that to generate more Aβ one needs to generate more APP, or make it easier for β-secretase to cut APP (As some mutations in APP do). Mutations in these genes are more rare, fAD is only like 5% of all AD cases, but having the mutations deterministically causes AD (Blacker & Tanzi, 1998).
After that, scientists looked for an explanation of how Aβ came out of APP. The currently accepted model is that there are two proteases (also termed in this case secretases), β-secretase (BACE1, and BACE2 gene) and γ-secretase, that cut APP to leave Aβ free. There are other secretases, most notably α-secretase (A number of different genes, but particularly ADAM10), and then a few other lesser known ones like η-secretase (Willen et al., 2015). The places in the protein where these make cuts were identified before the genes were, so initially all scientists knew was that "Something exists that has β-secretase activity". Multiple proteins could have such activity. There's a site the cut of which confers "ε-secretase" activity to that protein, but there is no protein that is directly called a ε-secretase. These secretases are not 100% specific and they can make cuts at different points. BACE1 can also cut at the β' site, which is also where BACE2 cuts and γ-secretase can cut at various γ-sites but also at the ε-site, conferring γ-secretase ε-secretase activity (to make things confusing). Of note, and potential interest later in the series of posts, a cut at the β' site, which lies in the middle of Aβ, would reduce the total amount of Aβ inside of the cell.
This picture which is often found in many papers, that APP sits in the membrane of the cell, as depicted by Herrup below is not quite accurate, but will get to that later.
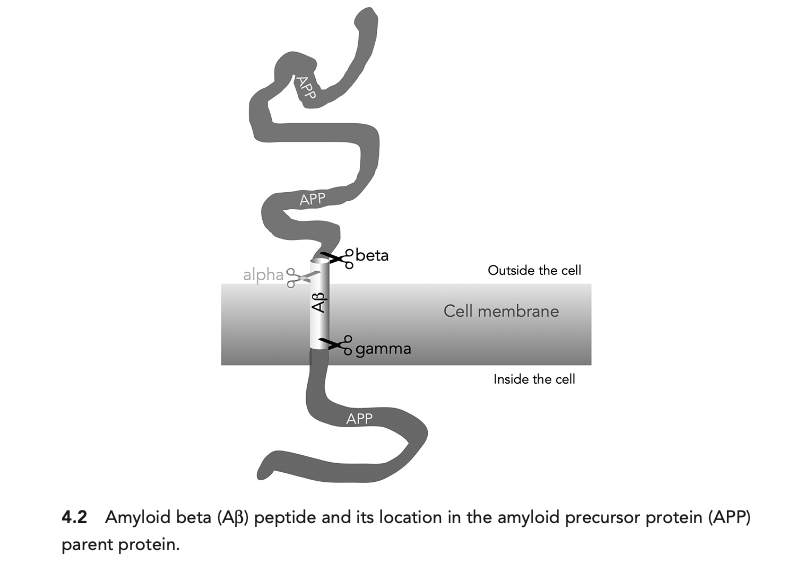
Eventually in the early 90s (1991, 1992) John Hardy for the first time coin the term amyloid cascade hypothesis in a form that has lasted until today. Literally in the 1991 paper's abstract: The pathological cascade for the disease process is most likely to be: β-amyloid deposition → tau phosphorylation and tangle formation → neuronal death.
The last scientific feat that cemented the ACH was engineering mice to express human APP. Mice by default don't develop Aβ plaques. Inducing hAPP led to plaques and some cognitive deficits. Removing the plaques (Using a vaccine to induce antibodies against Aβ) reversed the deficits (Schenk, 1999). On the basis of this discovery, a company called Elan started a clinical trial of the first ever therapy for Alzheimer's which was expected to be a success given all the mounting evidence. The drug, codenamed AN-1792 had substantial side effects and didn't slow down the deterioration of cognitive function, though it did remove Aβ plaques (Nicoll et al., 2003). That was 20 years ago, and since then many other monoclonal antibodies have been developed. Whether they worked in any meaningful sense is a debated topic which I will cover in a future post. Everyone was very confused: The genetic evidence seemed strong, and it seemed to work in mice. It cleared the plaques in humans. Why didn't it help the patients? That's not the topic of this post but we might return to it later. Suffice to say that this broad finding, the failure of immunotherapy against Aβ, is one of the two main pieces of evidence against the ACH, the other being the putative existence of patients that are free of cognitive impairment in the presence of lots of Aβ.
More recently, another approach was tested: inhibiting BACE1 (Das & Yan, 2020). This should lead to a reduction in the production of Aβ. With that, hopefully the brain could then clear the existing one, leading to a net reduction in Aβ, and with that, preventing cognitive decline. The first BACE1 inhibitor (CTS-21166) was tested in 2008. More recently others like lanabacestat (Astra Zeneca/Eli Lilly) and verubecestat (Merck) were also tested, leading to reduction in BACE1 activity, and reduction of CSF Aβ42. As you expect by now, no cognitive improvement either in advanced or early Alzheimer's (Egan et al., 2018; Egan et al., 2020). If you remember from the section above (and further explained in the Measuring amyloid section), higher CSF Aβ42 is what you see in healthy patients. BACE1 inhibitors do the opposite, they lower CSF Aβ42. More in a second.
Measuring brain function
What does "cognitively impaired" mean? In the context of Alzheimer's research a commonly used test is the Mini Mental State Examination (MMSE), but it is not the canonical, NIA-blessed test used for diagnosis. A lower score, particularly scores under 17 are interpreted as severe cognitive impairment. Another test is ADAS-cog, where scores higher than 12 tend to indicate dementia.
According to the NIA-AA guidelines, a diagnosis of Alzheimer's requires a clinical diagnosis of dementia. In turn, this diagnosis is given when :
Cognitively unimpaired Cognitive performance within expected range for that individual based on all available information. This may be based on clinical judgment and/or on cognitive test performance (which may or may not be based on comparison to normative data, with or without adjustments for age, education, occupation, sex, etc.). Cognitive performance may be in the impaired/abnormal range based on population norms, but performance is within the range expected for that individual. A subset of cognitively unimpaired individuals may report subjective cognitive decline and/or demonstrate subtle decline on serial cognitive testing. Mild cognitive impairment Cognitive performance below expected range for that individual based on all available information. This may be based on clinical judgment and/or on cognitive test performance (which may or may not be based on comparison to normative data with or without adjustments for age, education, occupation, sex, etc.). Cognitive performance is usually in the impaired/abnormal range based on population norms, but this is not required as long as the performance is below the range expected for that individual. In addition to evidence of cognitive impairment, evidence of decline in cognitive performance from baseline must also be present. This may be reported by the individual or by an observer (e.g., study partner) or observed by change on longitudinal cognitive testing/behavioral assessments or by a combination of these. May be characterized by cognitive presentations that are not primarily amnestic∗. Although cognitive impairment is the core clinical criteria, neurobehavioral disturbance may be a prominent feature of the clinical presentation†. Performs daily life activities independently, but cognitive difficulty may result in detectable but mild functional impact on the more complex activities of daily life, either self-reported or corroborated by a study partner. Dementia Substantial progressive cognitive impairment that affects several domains and/or neurobehavioral symptoms. May be reported by the individual or by an observer (e.g., study partner) or observed by change on longitudinal cognitive testing. Cognitive impairment and/or neurobehavioral symptoms result in clearly evident functional impact on daily life. No longer fully independent/requires assistance with daily life activities. This is the primary feature differentiating dementia from MCI. May be subdivided into mild, moderate, and severe
There's no official test that yields a diagnosis of dementia, it's up to individual clinicians to choose whatever tests they see fit for each patient.
In clinical trials of anti-Alzheimer's drugs, like Aducanumab's discontinued ENGAGE trial, Adas-Cog and MMSE were used as secondary outcome measures, whereas Clinical Dementia Rating Sum of Boxes (CDR-SB) was the primary measure.
In CDR-SB, scores of over 4 indicate mild dementia, and over 16, severe dementia (O'Bryant et al., 2008). This basically means taking this table, assigning a score in each domain, and then summing over all of them.
I will reuse this chart in a future post, for now you can look at what improvements have been achieved in the CDR-SB score relative to various amounts of reductions in amyloid PET signal (Haas & Selkoe, 2022).
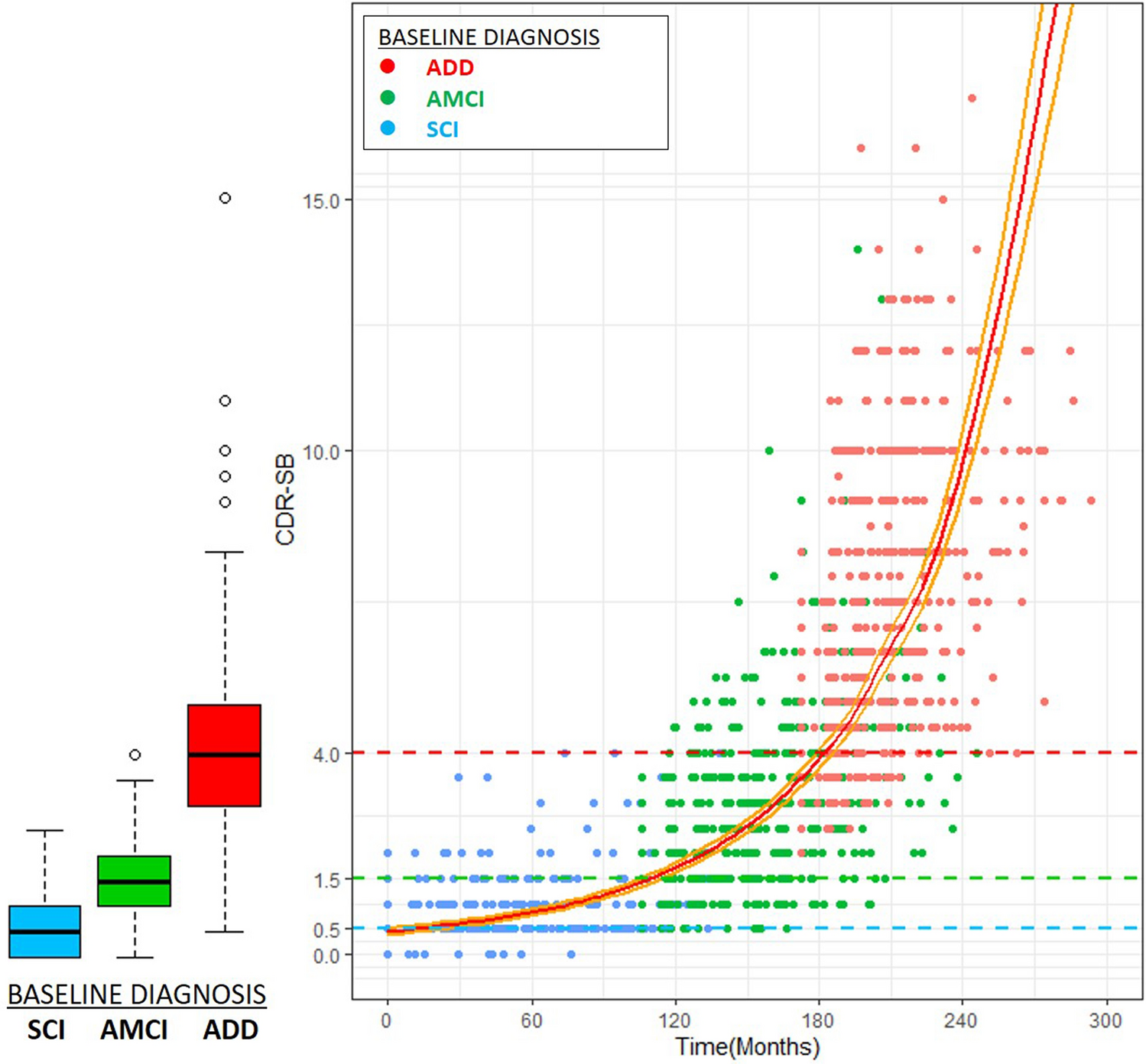
Here's (Kim et al., 2020) one chart that correlates CDR-SBs obtained a few years after an initial diagnosis of Subjective Cognitive Impairment all the way to AD:
Another reference, a large sample including patients with no diagnosis, mild cognitive impairment, and dementia, the average values are as follows (O'Bryant et al., 2010)
| Category | Age (avg) | CDR-SB (Avg) | MMSE (Avg) |
|---|---|---|---|
| Controls | 74.5 | 0.11 | 28.9 |
| MCI | 75.8 | 1.3 | 27.2 |
| Dementia | 76.6 | 7.22 | 19.2 |
| Probable AD | 77.5 | 7.4 | 18.8 |
What is Aβ?
We've been talking about Aβ/Aβ all this time without defining it precisely. Now it's time to fix that.
Amyloid beta (Aβ) is a peptide (a short protein) of 36-43 amino acids that is produced primarily in neurons as a result of the cleaving of a longer protein, the Amyloid Precursor Protein (APP) by β-secretase (BACE1 gene) and later γ-secretase (Itself a complex of proteins produced by genes NCSTN, PEN2, APH1, PSN1).
Aβ exists in multiple forms, mainly single peptides (monomers), clumps of it (oligomers), and finally fibrils of multiple chained Aβs (Figure from Kumar & Walter, 2011)
As discussed earlier, depending on what secretase is cutting APP where, there will be different kinds of Aβ formed. Aβ42 seems to be the one that aggregates into fibril whereas the second most common, Aβ40 is rapidly degraded. In mice, selectively expressing one or the other lets us see that only one leads to these plaques in vivo (McGowan et al., 2005).
Where is Aβ coming from? If you remember the figure earlier, with the secretases operating at the cell membrane, that's a common simplified model. In reality, β-secretase does not sit only at the cell membrane. It exists at many other membranes. It does not pop into existence at the cell membrane either. Rather, it is initially attached to endosomes inside of the cell, which then carry it outside of the cell. It is at these endosomes where Aβ is being produced, and this production and release can happen intracellularly.
What is Aβ for
When our bodies produce something that we suspect to be harmful, it is useful to think from an evolutionary point of view whether there is a reason to expect it would get made. Perhaps Aβ makes sense in a context that is not ours anymore. It could also be that some Aβ is useful and other isn't. Maybe Aβ-40 (non amyloidogenic) serves some function and Aβ-42 (amyloidogenic) is an accident. Given that Alzheimer's takes a very long time to develop, it's hard to think it would have been strongly selected against.
This all said, there is evidence that Aβ could be acting as an antimicrobial (First proposed, seemingly, by Robinson & Bishop 2002). A recent review of the evidence (Gosztyla et al., 2018) points out that:
- Interventions targeting Aβ have tended to cause more infections compared to the control group
- There are other proteins that we know what they are for, that also tend to aggregate in amyloids, like amelogenin (enamel in teeth)
- Aβ is relatively conserved across species. Long lived species like naked mole rats have very high concentrations of Aβ in their brains
- Some work has found that in AD patients, most of their plaques have HSV-1 (A common herpevirus) DNA whereas this is a much smaller amount for healthy people
- HSV-1 encephalitis correlates to lower levels of Aβ-42 in CSF
- In vitro, HSV-1 infection leads to intracellular Aβ accumulation
- The same is true in vivo, in mice. This is avoided by antiviral treatment.
- And it is also true for bacteria, in particular P. gingivalis (Hence the gingivitis-Alzheimer's correlation)
- Transgenic mice overexpressing APP survive markedly longer than regular mice when Salmonella is injected into their brains
I will leave for a later post an implication of this role of Aβ: that sporadic Alzheimer's is primarly caused by infections.
Other roles (Kent et al., 2020) hypothesised for Aβ are as a pseudo-platelet peptide, helping patch up the blood brain barrier (Which would then explain why removing Aβ from human brains sometimes leads to hemorrhage)
Measuring amyloid
Positron Emission Tomography
In humans, there are two main techniques used to look for amyloid: PET-Aβ and histopathology (sectioning and staining the brain with a dye that reacts with amyloid, like Congo red). Are these good?
PET techniques give the patients compounds, mainly Pittsburgh Compound B (PiB) or Florbetapir. These bind to Aβ and allow clinicians to see its accumulation in the brain using PET. From the wiki article:
11C-PiB is a fluorescent derivative of thioflavin T which preferentially targets and binds to fibrillar Aβ forms found in dense core plaques with high affinity and specificity. In particular, it specifically binds to Aβ40 and Aβ42 fibrils and insoluble plaques containing the aforementioned Aß peptides. PiB does not bind with great affinity to soluble or nonfibrillar Aß plaques until plaques have reached a crucial magnitude, which has yet to be determined.[3] Furthermore, this radiotracer does not bind to neurofibrillary tangles (NFTs) in the neuronal regions of the brain during postmortem autopsies
Alternatively, one can try to measure glucose consumption in the brain: With Alzheimer's neurons die so there's less use for the glucose. fluoro-2-deoxy-D-glucose (FDG) can be used to this end. Importantly, FDG measures something that's downstream of amyloid, glucose metabolism: This is important because it allows one to bypass the potential critique that the amyloid that matters is not being measured by PET-FiB.
This latter technique is one that should yield results comparable to looking at brain volume directly by MRI. Do these correlate with cognitive functioning scores?
PET-FDG or PET-Florbetapir correlate somewhat with cognitive function, but FDG being a measure of how many neurons there are, it makes sense that it correlates better with function (Khosravi et al. 2019):
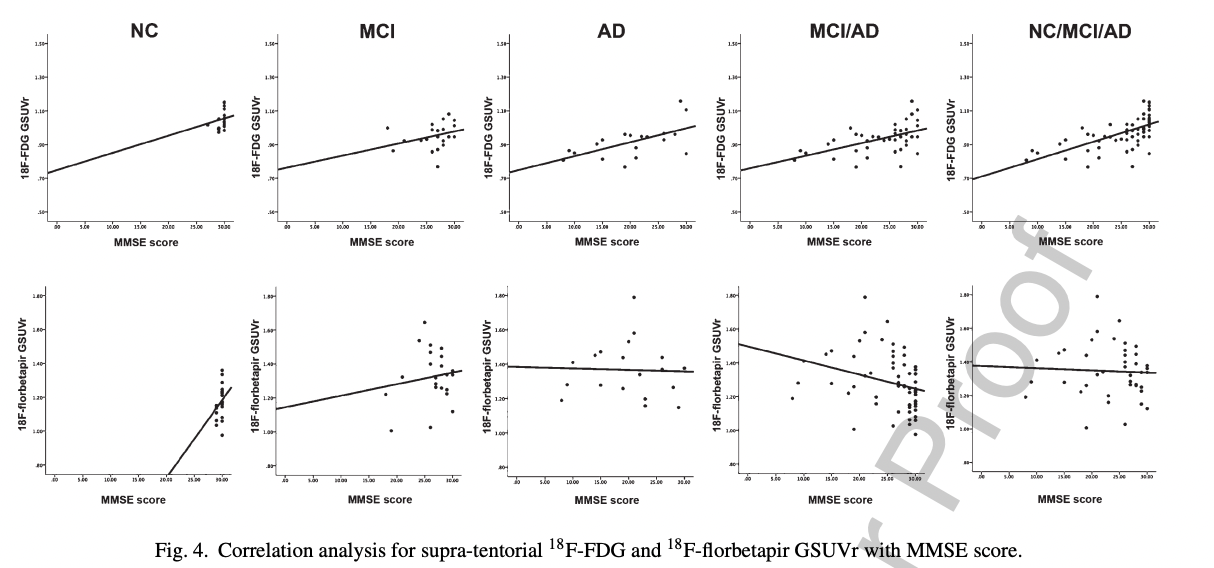
That FDG better correlates with function is unsurprising: It is causally closer to the variable of interest (cognitive function): here are many reasons why someone's neurons might be dying besides amyloid!
PET does not measure all amyloid, the NIA-AA article warns that a negative amyloid PET scan should not be equated with the complete absence of Aβ in the brain or even with absent or sparse neuritic plaques
CSF Aβ42 & Aβ42/Aβ40 ratio
Yet another way is to measure presence of Aβ in the cerebrospinal fluid (CSF). In particular, one could measure Aβ-42 or the Aβ-42/Aβ-40 ratio. The idea here is that Aβ-42 is the form of the peptide that tends to aggregate. In later stages of the disease, the plaques are seemingly acting as sinks for it, and one observes Aβ-42 going down (more on this in a bit). However, different patients produce Aβ-42 at different rates, so one has to normalize by Aβ-40. This ratio is a much better predictor of cognitive impairment.
Some key papers:
Niklas Mattsson et al. (2014)
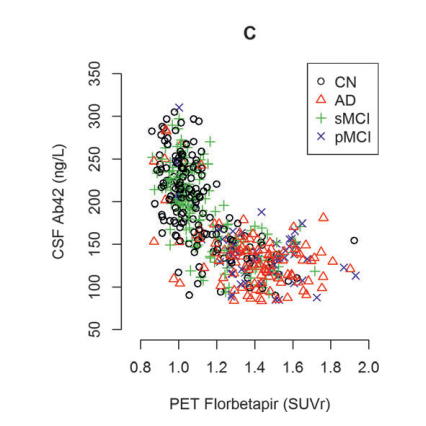
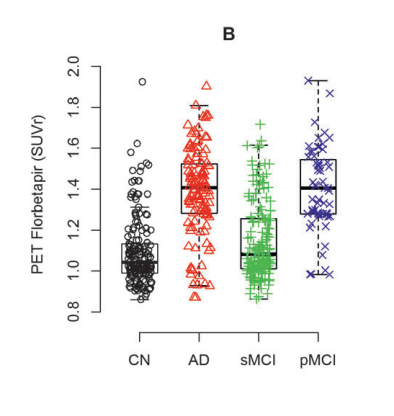
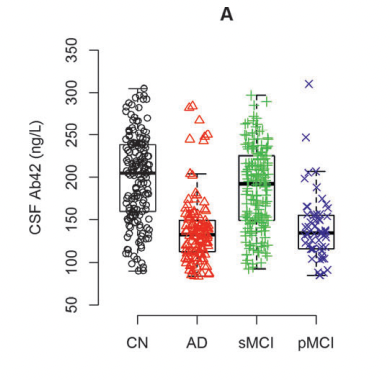
In these figures, MCI is stable mild cognitive impairment and pMCI is its progressing form (which could end up in Alzheimer's). AD is Alzheimer's and CN is normal patients. Note also that there does seem to be a number of normal patients that are deep in the "low Aβ42 high PET" group. PET seems to have higher specificity: A high SUVr is a better predictor of Alzheimer's than is a low Aβ42 value in CSF.
As a biomarker, Aβ-42 can be improved if one normalizes it by Aβ-40 (which remains unchanged with aging). This is what Baiardi et al. (2018) do, showing a tighter correlation with amyloid pathology. This Aβ score below was obtained by slicing FFPE-fixed brains and staining them with antibodies against Aβ, directly measuring how much amyloid there was in the brain. This being antibodies, it means they are measuring largely extracellular amyloid.
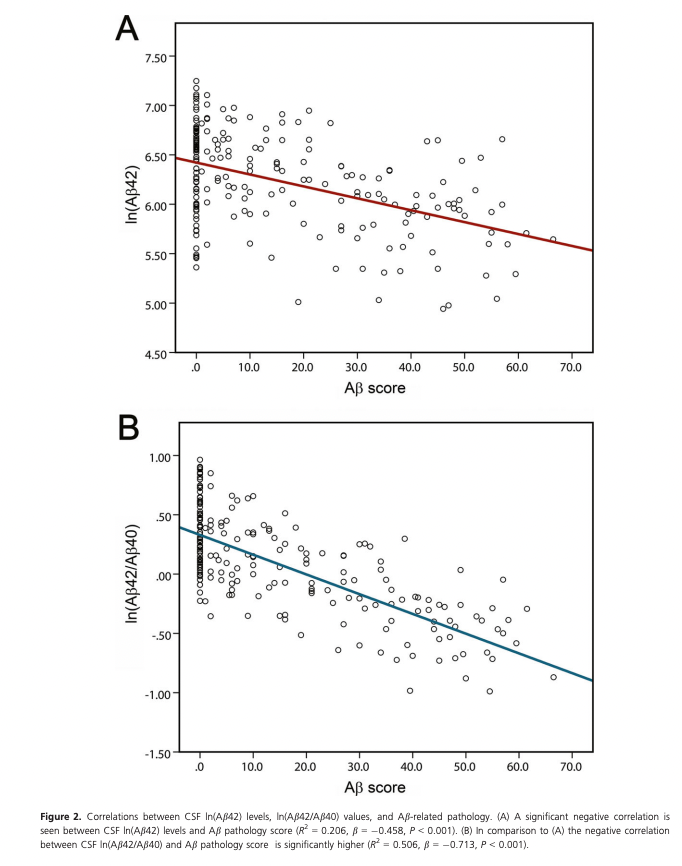
Using antibodies is an imperfect way to look at amyloids because there are many other Aβ peptides that may not be problematic, the 4G8 antibody used in this paper, for example also reacts with other shorter Aβ fragments as well as the P3 peptide, which has a sequence overlap with Aβ4. (Hunter & Brayne, 2017).
Do these correlate with brain function?
It seems so: Less Aβ42 in your CSF, worse cognitive function (Bousiges et al., 2016):
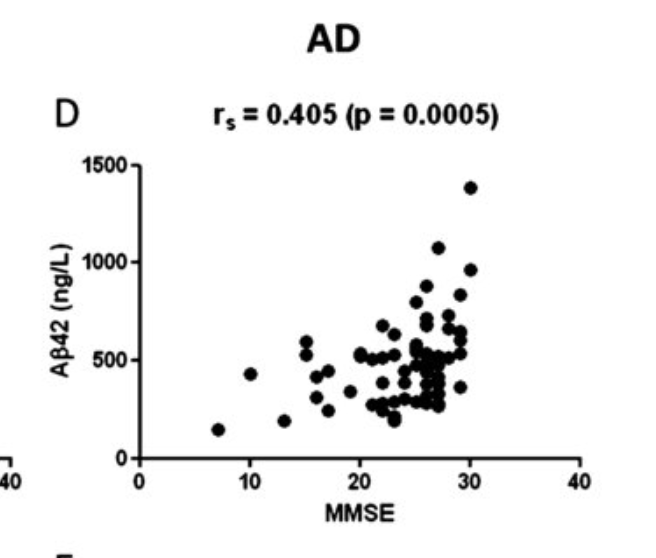
It seems surprising that lower Aβ42 means lower function, especially given the fact that BACE inhibitors or antibodies against Aβ reduce CSF Aβ42. CSF Aβ42 does not seem to change with age (unless you get Alzheimer's), as opposed to tau, which does increase with age (Sjögren et al., 2001).
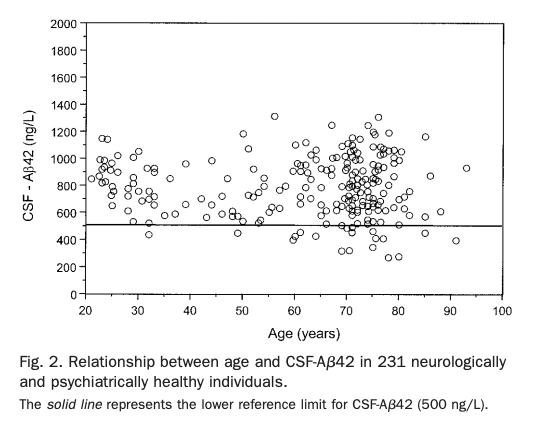
Spies et al. (2012) look into this, summarizing various arguments for where this decrease is coming from:
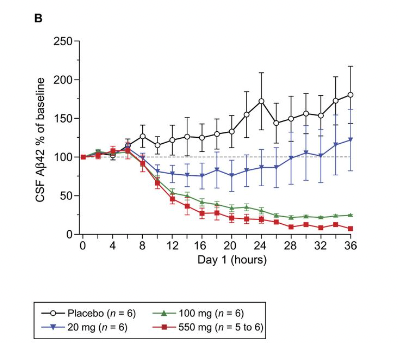
They ultimately think that it is indeed Aβ42 aggregating in plaques that is acting as a sink for the Aβ. But if the plaques are being cleared and no more Aβ42 is being produced, then the Aβ42 from the plaques should be getting to the CSF as it is being cleared, raising it. Then it should go down as no more is being produced. Maybe this is what happens, but very fast? This is from a single injection (Kennedy et al., 2016), there is a slight increase a few hours post and then a decrease, but the same seems true of the placebo:
So I guess what must be going on is that the rate of clearance from brain to CSF is constant, so Aβ42 is removed from the brain at the same rate as usual, until there is no more Aβ42 (because BACE has stopped making it), and then it drops. This graph here is in healthy volunteers, but something similar can be appreciated in AD patients:
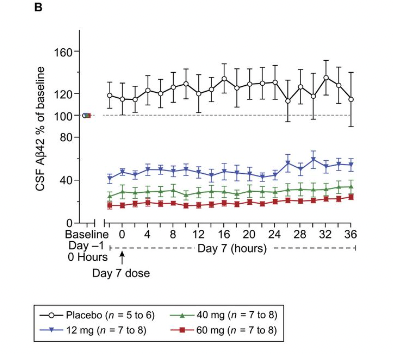
This suggests that even in the elderly with AD, the brain can clear amyloid plaques if there's no additional amyloid being added.
What is tau?
I've been talking above mostly about Aβ. But in the ACH it is tau, and not Aβ, that is the proximate cause of neurodegeneration.
Tau is a protein encoded by the MAPT gene (Avila et al., 2004). Tau proteins bind to microtubules (cellular scaffolds) to modulate their stiffness. When tau is phosphorylated (perhaps by kinase GSK3 or CDK5), the protein falls off from microtubules (cellular stiffness goes down) and that freed up tau clumps with other taus into fibers. MAPT is mostly expressed in neurons and glials cells, as well as sperm precursor cells (spermatids).
Once tau falls off it first forms paired helical fibers (PHFs) and then those get entangled into neurofibrillary tangles (NFTs)

Given that tau is bound to microtubules inside neurons, it's no surprise that these form inside of the cell first, but they can also be found outside, released once a cell dies, but biology being biology, it's also possible that individual tau proteins get secreted and then PHFs and later NFTs form outside of the cell as well, or perhaps some cells end up with intracellular NFTs due to endocytosing extracellular ones (Avila, 2010).
The specific sequence of events leading to neuron death seems to involve a buildup in intracellular tau followed by cell death, leading to a leak of tau outside of the cell, then causing other neighboring cells to die and release their tau in turn in a positive feedback loops that leads to tau pathology spreading through the brain. The concentration of tau that is hypothesised to exist around a dead neuron in vivo is within the toxicity range of tau that was found in vitro (Gomez-Ramos et al., 2006).
Given the fact that tau pathology does not spread randomly through the brain but through connected regions of the brain, it makes sense to consider the possibility that NFTs are prion-like in nature and induce further aggregation of proteins in close proximity (Mrdjen et al., 2019):
NFT accumulation begins in the axons of neurons where normal tau is present. Pathologic tau then becomes mislocalized from axons to cell bodies and dendrites by a thus far unknown mechanism, where it mediates synaptic dysfunction independently of neuronal degeneration [77]. Trans-synaptic spread of tau aggregates in a prion-like manner has been investigated extensively in experimental systems, where initial aggregated tau seeds the spread of pathologic tau through synaptically connected regions [65, 168, 180]. Clavaguera et al. have shown that injection of brain homogenates from human patients with different tauopathies into a mouse model that expressed full-length human tau resulted in near replication of regional patterns of neuropathologic changes seen in the human source [33]. Similarly, Boluda et al. demonstrated rapid and region-specific spread of pathological tau following injections of AD brain homogenates in transgenic mice [11]. Brain accumulation of pathologic tau can be induced in mice by intracerebral or intraperitoneal injections [11, 33, 34], and pathologic tau propagation is enhanced through increased neuronal activity [193, 195]. One consequence of these and similar experimental demonstrations of pathologic protein transmission in animal models has been the proposal that AD may be transmitted in a prion-like manner [19]. Analogous to the spread of prion protein, intracellular tau may be transferred from neuron-to-neuron via physical connections like tunneling nanotubes [172]. An alternative proposition is that tau could be released into the extracellular matrix within secreted extracellular vesicles like exosomes or microparticles [137, 156, 165]. Indeed, exosomes containing hyperphosphorylated tau are released upon depolarization of neurons in vitro [183].
Experimentally, one can inject misfolded human tau into wild-type mouse brains and cause murine tau to start to aggregate and reproduce the pathological cascade seen in humans (Soto & Pritzkow 2018; Vaquer-Alicea et al., 2021; Narasimhan et al., 2017)
Tau has multiple conformations (it's an intrinsically disordered protein) and aggregates in different ways: The kind of tau aggregates one sees in AD is the same as in Primary Age-Related Tauopathy (PART), but in turn that's different from what one sees in tauopathies derived from traumatic brain injury or Pick's disease (Shi et al., 2021)
The topography of Alzheimer's
Progression of NFT and Aβ pathology through the brain does not follow the same pattern. Amyloid deposition starts in the neocortex and then makes its way towards the center of the brain, following the Thal stages:

Whereas tau starts spreading before Aβ appears from the Locus coeruleus:

Over time, the evolution of Alzheimer's biomakers looks like this (Zetterberg et al., 2020):
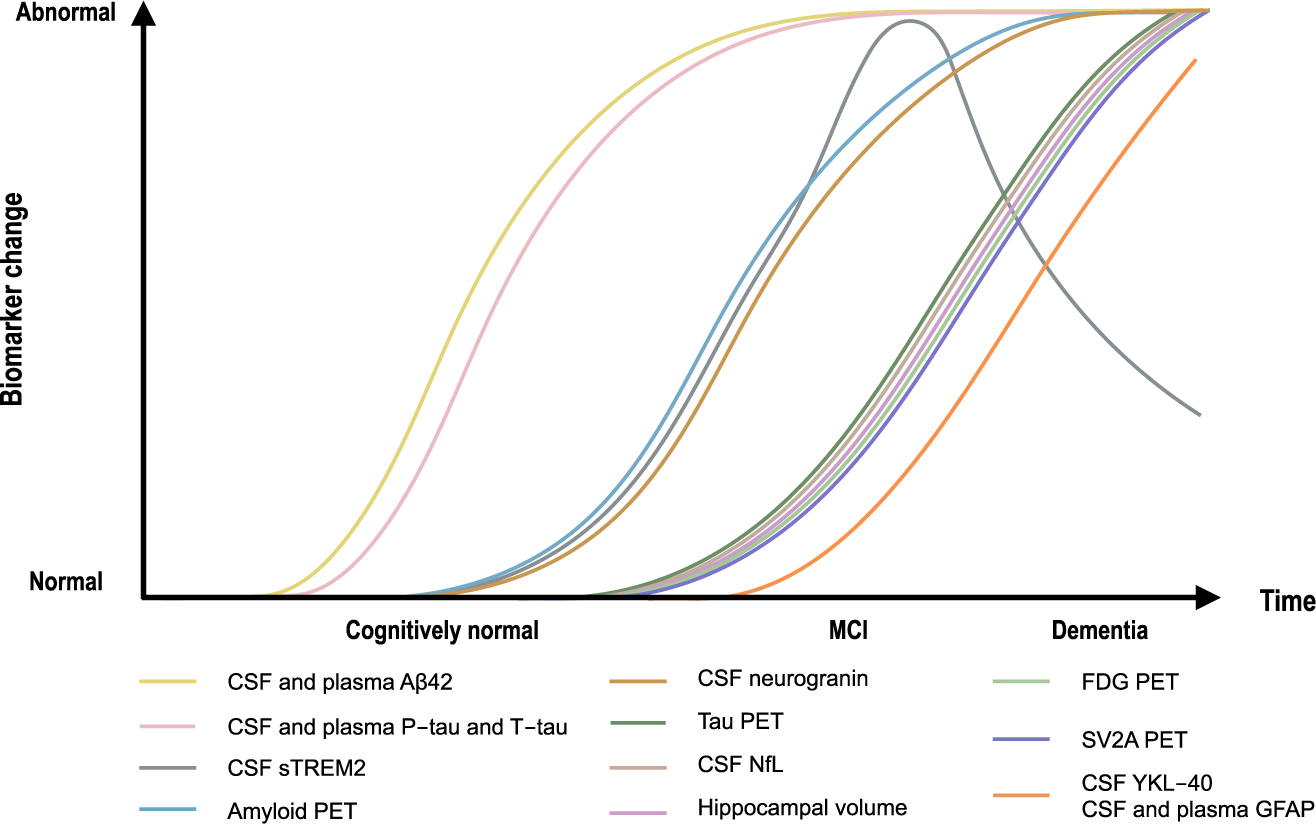
Locus coeruleus, the root of all evil?
In the canonical ACH model as originally proposed, tau pathology follows Aβ plaque deposition. However, most people have some NFTs in their brains way before they get any amyloid, and this initial NFT formation starts at the locus coeruleus (LC), hence a better reframing of the ACH is that per Braak et al. (2011)
Sporadic AD may be the result of two separate assaults: first, a tauopathy, possibly beginning in childhood; and second, negative influences of β-amyloid after a given threshold is crossed. β-amyloid might be capable of exacerbating the underlying tauopathy so that it develops into clinical AD (69-71). If the pretangle material is not regressive or transient (72), our findings may indicate that the pathologic process leading to abnormal tau pathology and ultimately capable of inducing NFT formation does not begin in the transentorhinal region but in select subcortical nuclei; it may commence before puberty or in early young adulthood. Currently, too little is known about the pace at which the pathologic process develops. Some individuals were still at NFT Stages I and II at 90 years and older (Fig. 2A). Thus, although all individuals in this study who were 40 years and older exhibited pretangles (owing to a considerable variability in the rate of progression), only a proportion of them would have gone on to develop AD had they lived longer.
If other groups replicate our present findings and confirm that the AD-related tauopathy begins in noradrenergic projection neurons of the locus coeruleus, it might be possible to intervene with therapeutic means earlier than at present, that is, during the first decades of life, by protecting caeruleus projection cells and/or preventing them from developing the pretangle material (73-75).
One can then try to think about what makes the LC special. One hypothesis is that it is particularly exposed to external toxins or viruses due to its location (Satoh & Ijima, 2017):
In addition, LC neurons are highly exposed to blood circulation and cerebrospinal fluid. The LC innervates the great majority of the CNS microvasculature and its associated astrocytic endfeet (Cohen et al., 1997). According to the total number of LC neurons (32,000) (Aston-Jones and Cohen, 2005) and estimated length of capillaries (640 kilometeres) (Pardridge, 1999) in human brains, each LC neuron innervates 20 meters of capillaries on average. No other neurons have such widespread contact with circulating blood (Pamphlett, 2014). Thus, with their large exposure to the blood circulation, LC neurons could take up toxins even if they were at low levels in the blood, and this risk would increase when the integrity of the blood-brain barrier is disrupted under diseased conditions, such as AD (Kisler et al., 2017). In addition, the close proximity of the LC to the forth ventricle may also expose it to toxins in cerebrospinal fluid (Mravec et al., 2014).
Taken together, LC neurons are faced with the risk of taking in harmful metabolites, toxins, and even viruses (Braak and Del Tredici, 2015; Mravec et al., 2014; Pamphlett, 2014), which may cause oxidative stresses, metabolic challenges, and aberrant immune responses. These stressors may exacerbate tau aggregation and toxicity, and increase the risk of LC neurons to develop NFT pathology and neurodegeneration during aging. Therefore the LC may be an effective site to target in research to prevent or reduce brain aging, as well as AD.
Another is metabolic stress due to inefficient neuron to neuron communication due to poor myelination
One hypothesis that could confer this susceptibility is the cytoarchitecture of these cells, as LC neurons have long and thin axons that have poor or incomplete myelination [153]. Such a characteristic forces the cell to increase its energy output to perform efficient action potentials that lead to increased cellular oxidative stress, and gaps in the myelin sheath allow greater exposure to environmental toxins (Matchett et al., 2021)
In patients that end up presenting AD, reduction in volume of the LC due to neuronal deaths happens early in life, but there is no such reduction in volume in the elderly with normal cognitive function (Matchett et al., 2021).
Previous studies have suggested that LC degeneration is age related [50, 85, 93, 157, 164]. However, it is of note that some of these findings were based on a small cohort (n = 5–13) [50, 85] and did not exclude cases with neuropathology elsewhere. Intriguingly, recent unbiased stereological approaches indicated that there are no significant associations between normal aging and changes in the LC [16, 106, 116, 154], suggesting a disease-specific phenomenon.
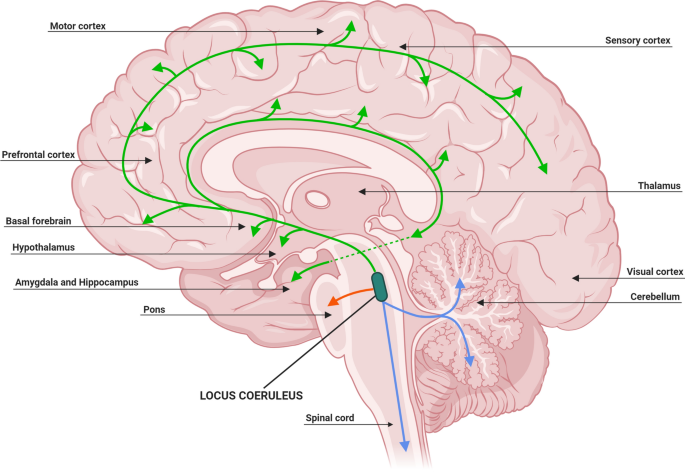
The explanation for why tau and Aβ start where they do I have given is the same "higher metabolic activity" plus "exposure to external toxina and pathogens" (for the LC). To give a full, satisfying account, of the observed pattern we would probably have to go into a longer discussion of specific types of neurons that are more or less vulnerable to Aβ or tau. I found an article that discusses that here (Mrdjen et al., 2019). Apparently the most resistant neurons to tau accumulation are the Purkinje cells in the cerebellum and the most vulnerable ones, glutamatergic [glutamate producing] excitatory neurons.
Different parts of the LC are connected to different parts of the cortex. Whereas NFT accumulation starts in the LC, amyloid accumulation starts in areas that have been singled out as the default mode network (Knopman et al., 2021), perhaps because they are under higher stress. Generally, there seems to be a correlation between how well connected an area of the brain is and how much Aβ plaque it will get (Buckner et al., 2009)
Amyloid and tau co-causing Alzheimer's
In aged patients without Alzheimer's, NFTs are found in a limited number of brain regions. Once tau makes it to the cortex then those individuals go on to develop AD (Musiet & Holtzman, 2015)
In sAD, the relationship between Aβ and tau pathology is complex, but ultimately supportive of the amyloid hypothesis. Aggregated, phosphorylated tau pathology is present in the brainstem and entorhinal cortex of young, normal, asymptomatic people and precedes the onset of amyloid accumulation38. With age, hippocampal neurofibrillary tau pathology is nearly ubiquitous, but remains confined to limbic regions in cognitively normal, amyloid-free individuals39,40. However, tau appears to spread into neocortical regions almost solely in people with coexistent Aβ pathology39,41. These individuals generally go on to develop AD dementia. In several studies examining the cognitively normal elderly or patients with mild cognitive impairment or AD, widespread cortical tau pathology (Braak stage ≥3) was commonly observed in patients with concomitant Aβ plaques; it was never observed in those without plaques, suggesting that the presence of Aβ aggregation is required for the appearance of high-grade cortical tau pathology39,41,42. Furthermore, although hippocampal tau phosphorylation is commonly observed in cognitively normal adults, studies have found no evidence of neuronal loss in these regions with normal aging43,44. However, neurodegeneration does occur prominently in these regions in AD patients with amyloid plaques43,44, suggesting that tau-mediated toxicity again requires some trigger from Aβ. Accordingly, 'neuritic plaques', which are amyloid plaques with associated neurofibrillary tau pathology, correlate more closely with neuronal loss and dementia in AD than either plaques or tangles alone45. These neuropathologic observations support the idea that while fibrillar Aβ and neurofibrillary tau pathologies are anatomically separated, Aβ aggregation appears to somehow result in accelerated neurofibrillary tau pathology and neuronal toxicity. As noted above, fAD mutations, which directly influence Aβ aggregation, cause tau pathology that is similar to that seen in sAD and correlates closely with neuronal death. However, although mutations in the gene encoding tau (Mapt) cause neurodegenerative disease, they do not induce Aβ pathology46,47. Accordingly, polymorphisms in Mapt are associated with increased CSF phospho-tau levels, but only in patients with CSF evidence of Aβ pathology48. Thus, human neuropathological data supports a key tenet of the amyloid hypothesis: that Aβ triggers or exacerbates downstream tau pathology, although it appears to do so without anatomic colocalization.
This may not be as true in familial Alzheimer's (fAD), where excess production of net Aβ leads to amyloid plaques appearing first. Intriguingly, another common finding is that familiar Alzheimer's starts earlier, but it does not progress faster: the presence of tau seems to be rate limiting and its spread seems to be relatively independent of Aβ. This speaks in favor of a model where Aβ plaque accumulation starts the engine, so to speak, but then tau-induced neurodegeneration may be self-sustaining. If so, trying to remove amyloid beta from patients where AD is already ongoing will achieve little, one would need to remove tau or perhaps tau and Aβ at the same time.
There is some evidence than on its own tau does not lead to Alzheimer's (That is, to the appearence of amyloid plaques)
mutations in the gene encoding tau (Mapt) cause neurodegenerative disease, they do not induce Aβ pathology46,47. Accordingly, polymorphisms in Mapt are associated with increased CSF phospho-tau levels, but only in patients with CSF evidence of Aβ pathology48. Thus, human neuropathological data supports a key tenet of the amyloid hypothesis: that Aβ triggers or exacerbates downstream tau pathology, although it appears to do so without anatomic colocalization. (Musiet & Holtzman, 2015)
Additionally, people with the so called Icelandic mutation (or A673T, an alanine to threonine replacement at position 673 in APP) don't develop Alzheimer's, perhaps because the conformation of APP resulting from the mutation makes BACE1 cut preferentially at the β′ site, which is not amyloidogenic (Jonsson et al., 2012, Guyon et al., 2020, Kimura et al., 2016). These people do seem to have some NFTs in their brains, but without the amyloid "spark" they don't lead to major problems. If only we had perfected gene therapy delivery through the brain, we could just insert this one mutation in all neurons and prevent AD from ever starting (Tremblay et al., 2022)
So what is happening in the AD brain? What happens when Aβ and tau get together?
(A->T)->N?
There isn't much evidence in vivo in humans about how exactly Aβ and tau work together to cause neurotoxicity. But there is some in mice and in vitro. In mice, it's possible to do things like inject Aβ to a mouse that overexpresses tau (but not Aβ) and see if one sees more NFTs near the injection sites, which is indeed what happens.Bloom (2014) has a summary of these studies:
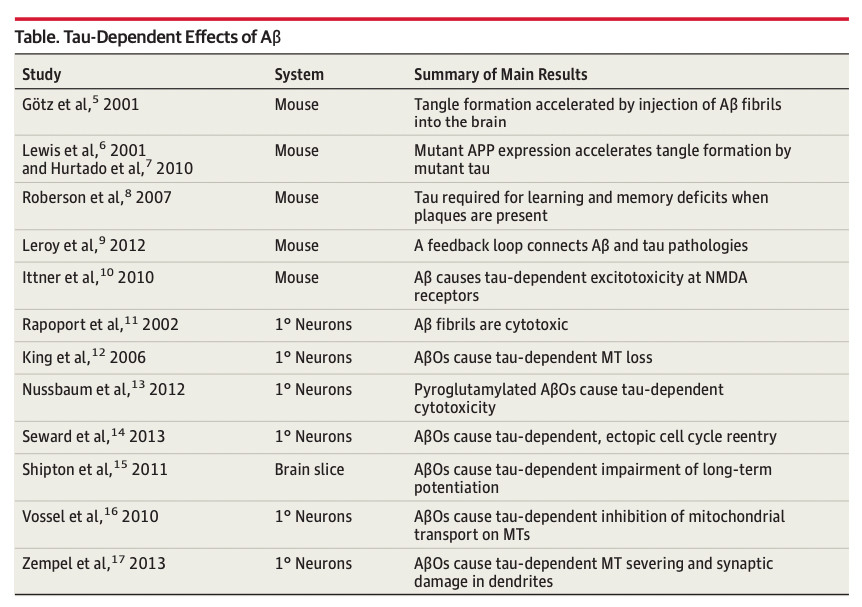
On its own, without tau, Aβ does not seem to lead to much in humans. Perhaps it leads to some mild cognitive deficits which can be reversed by clearing Aβ, which is what we see in mice. Once tau comes into play maybe Aβ ends up also being toxic? One implication of this is that if you are working with an Alzheimer's mouse model and you see an improvement in a cognitive assay, you are on the wrong path, you should not be able to recover a true AD phenotype, because that involves neurons dying (which are unlikely to recover, at least not in the short term), what these mouse models are probably showing is that the impaired cognition resulting from Aβ plaques can be reversed. Earlier in the measuring amyloid section I had a chart of amyloid plaque reduction correlating with some cognitive improvement which would make sense under a model where two pathologies coexist, minor cognitive impairment due to Aβ plaques, and the actually destructive AD which current therapies do not reverse.
Evidence that tau pathology is not just an epiphenomenon of Aβ pathology, but instead that tau is required for Aβ toxicity in vivo, emerged from crossing tau knockout mice with hAPPJ20 mice that overexpresshumanAPPcontaining2mutations,eitherofwhichcauses familialearly-onsetAD on its own. Plaqueaccumulation in hybridmice that were either tau null or contained 1 tau gene was identical to the parental APP strain that contained 2 tau genes. Remarkably, loss of either 1 or 2 tau genes protected hybrids against the learning and memory deficits and excitotoxicity characteristic of the parental APP strain.8 These results imply that Aβ initiates a pathway that leads to tau-dependent synaptic dysfunction. Moreover, they raise the possibility that the cognitive and memory loss associated with AD can be prevented or decelerated by reducing the level of tau in the brain.
Busche & Hyman (2020) have a more recent review of the evidence, concluding similarly that the Ab-tau interaction is key to AD but not yet well understood:
compelling experimental and clinical data have emerged that support a co-pathogenic interaction between Aβ and tau in AD, which not only manifests throughout the disease course, but also fundamentally drives disease progression. It is therefore tempting to speculate that reconceptualizing AD in terms of Aβ–tau synergy may provide a vital therapeutic context (Box 1) in which to rationalize past failures and inspire future successes. Nevertheless, it is clear that our knowledge of Aβ–tau synergy remains in its infancy, with several outstanding unknowns, including the etiology of within-model inconsistencies or the contributory role(s) of other critical processes, such as glial activation, that must be explained before the complexities of a decades-long evolution of neurodegeneration can be modeled under such a framework.
Conclusion
This post contains a summary of the consensus view on Alzheimer's, along with some ancillary tidbits required to understand how AD is diagnosed and measured. Alzheimer's is caused by a combination of Aβ and tau, leading to neurodegeneration and loss of cognitive function. The data seems compatible with some form of the ACH, but not the most naive form. It seems that rather than amyloid sparking tau on its own, tau pathology antecedes amyloid beta plaque deposition, and once the disease has started, it seems to be self-sustaining and independent of amyloid.
I have not examined in any depth some alternative explanations for AD like infections, or loss in glymphatic system function. I will do so in following posts. I haven't explained in detail what the arguments against the ACH are, and again I will do so in a future post.
Appendix: An alternative way to produce Aβ without BACE1?
At the beginning of the Alzheimer's story it was unclear how Aβ got made. There was a competing hypothesis to the existence of a protease cleaving APP: Right in the middle of APP, at the beginning of the Aβ section (Position 671) there is a methionine amino acid. One that is surrounded by a pattern in nucleic acids known as the Kozak sequence. This sequence tends to appear on sites that are sites of protein translation initiation. That is, the mRNA gets made, and then the ribosome starts making protein starting from there. What if instead of APP getting cleaved at that spot, translation starts at that point directly? In that case there is no need for BACE1 to cut it! This was indeed proposed by Breimer & Denny (1987):
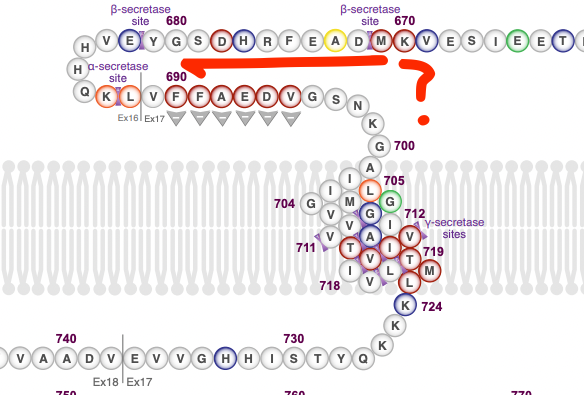
They note that if this were to happen, then the resulting protein would lack the initial sequence, which codes for, among other things, a signal peptide right at the beginning of it (N-terminus) that homes the protein to cell membranes. This protein would then have no "reason" to get outside of the cell, eventually leading of Aβ buildup intracellularly. This theory has been tested a number of times: The putative protein that results from internal translation was not found in brain homogenates from AD patients (Dyrks et al., 1988).
Macq et al. (1994) expressed the sequence in Chinese Hamster Ovary (CHO) cells starting at the methionine to see if that would get translated and it does (with reduced efficiency compared to the fill APP). Then, to see if the full length APP mRNA could give rise to the truncated protein, they inserted a stop codon upstream of the methionine: This way a ribosome translating APP would stop there, before getting to the Aβ section, and all produced Aβ must then be coming from alternate translation. No Aβ peptide was detected in these conditions though. Similar findings were reported by Citron et al. (1993) in human kidney cells.
More recently, Volloch & Rits-Volloch (2022) have pointed out that maybe this mechanism would still work but only under specific conditions, namely in human neurons (Macq and Citron tested in different cells), and under stress (specifically when the Integrated Stress Response (ISR) is active). This specifically has not so far been tested experimentally.
What has been tested is whether APP can be translated in a cap-independent way, at an Internal Ribosomal Entry Site (IRES) which tend to be active under the ISR (Bond et al., 2020, Beaudoin et al., 2008). It can. Under the ISR global protein synthesis rates go down and IRES/cap-independent translation occurs for a small subset of proteins. One of which is APP. So who knows! Someone should probably test this in human neurons that are stressed out and see if when blocking BACE1 one still sees amyloid being produced. I did email Martin Citron who confirmed no one has looked at neurons, and that while this alternative source of Aβ has a low likelihood, it hasn't been directly ruled out.
Acknowledgements
To Willy Chertman, Aβmale who shared pointers to his own summary of Alzheimer's here and here, and everyone that replied to this Twitter thread. Marti JM.
Changelog
- 2022-10-17: Typos. Bousiges et al. did not measure Ab42 in blood, it was CSF.
Citation
In academic work, please cite this essay as:
Ricón, José Luis, “An introduction to Alzheimer's Disease”, Nintil (2022-10-13), available at https://nintil.com/what-is-alzheimers/.
Comments