The failure of monoclonal antibody therapy for Alzheimer's Disease, implications for the Amyloid Cascade Hypothesis
Table of Contents
In a previous post I explained the basics of Alzheimer's disease and the current state of the art in our understanding of it. I focused on the amyloid-tau story while hinting at the fact that there are other factors worth exploring. I mentioned the "repeated failures of Alzheimer's drugs in the clinic". This post is about that.
I mostly focused on the Aducanumab case, with solanezumab and lecanemab as contrasts.
These trials have been used as evidence against the amyloid cascade hypothesis (ACH). After reviewing them I do not think these refute the ACH, but they strongly speak against a naive model where amyloid removal at any stage of the disease will stop progression.
Aducanumab
Aducanumab/Aduhelm is a monoclonal antibody against amyloid that was controversially approved by the FDA despite their Peripheral and Central Nervous System Drugs Advisory Committee recommending not approving it (None of the 11 members recommended approving it) . Some of them even resigned over the FDA's decision.
Many were worried that aducanumab, assuming it indeed doesn't work, would cause doctors to prescribe it, raising aggregate healthcare costs in the US for no benefit. Fortunately (for the US economy) and unfortunately (For Biogen and Eisai, developers of the drug), aducanumab has not seen much adoption.
Aducanumab targets aggregated forms of amyloid in particular: not every monoclonal antibody is the same:
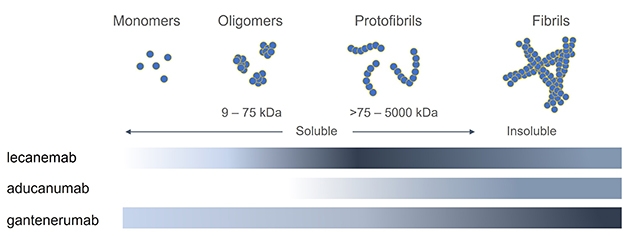
Aducanumab was first tested in humans in the PRIME trial Phase Ib in 2016 and multiple other tests have evaluated its efficacy since then (ENGAGE, EMERGE, PROPEL) in various patients populations, from healthy to probable AD. The largest of these were ENGAGE and EMERGE, on ~1600 patients across the world with MCI or mild AD. Two larger trials, EMBARK (2400 participants) and ADUHELM ICARE AD-US (6000 patients) are ongoing.
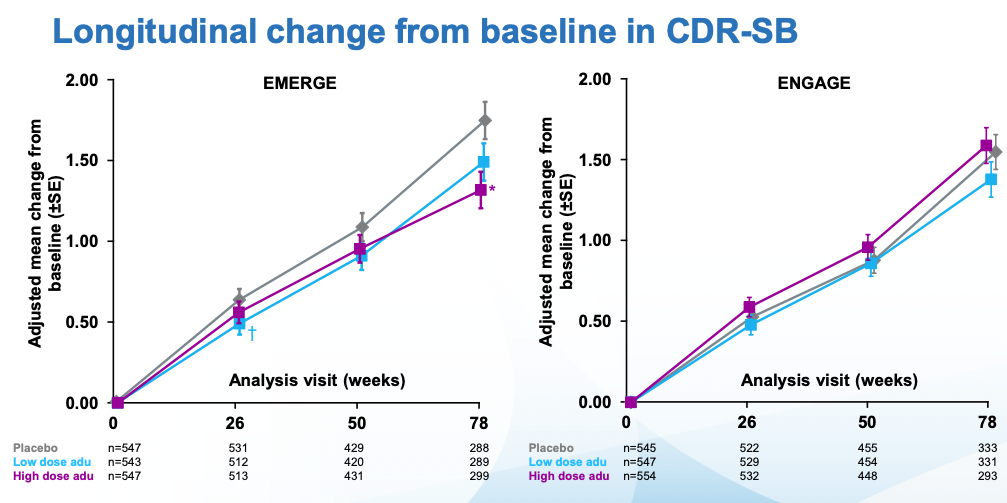
The doses in these two similar larger studies (ENGAGE and EMERGE) went up to 10 mg/kg, dosed once a month. Everyone was followed for 18 months. The main outcomes to look at for these trials are amyloid PET (to see whether amyloid was indeed removed) and CDR-SB, a measure of cognitive function I review in my previous post. As a reminder a CDR-SB=0 means good cognitive function. The patients in this study started with CDR-SB~2.5 which qualifies as MCI. CDR-SBs over 4 would start to get into AD territory. In the undiagnosed population of similar age (~74), CDR-SB is ~0.11. With that said, here are the topline results for both trials per Biogen:
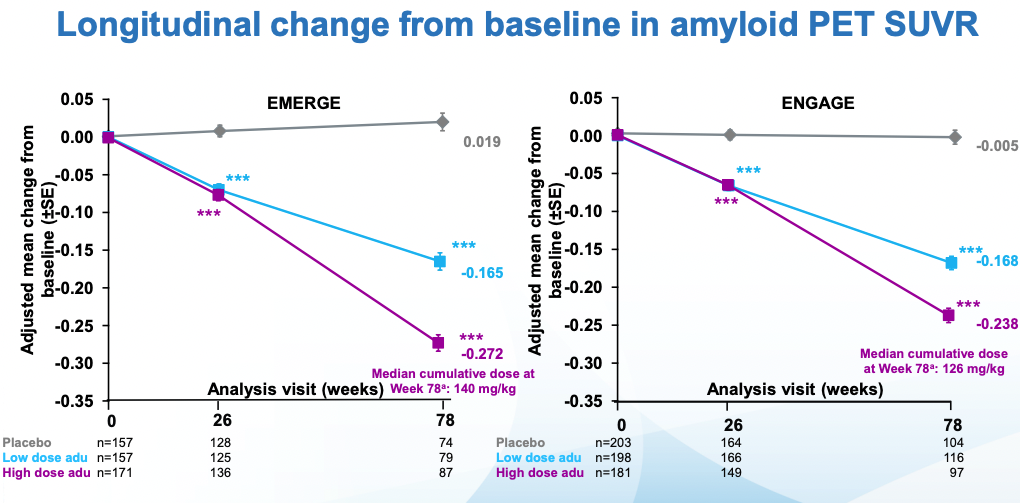
Amyloid reduction looks good: a dose-dependent reduction in Ab PET signal. The initial signal was ~1.37 so one can take this to mean a 20% reduction in amyloid in the high dose case. The values here are not the raw values, rather the fine print in the slides I'm citing notes that a number of reasonable-sounding corrections were made.
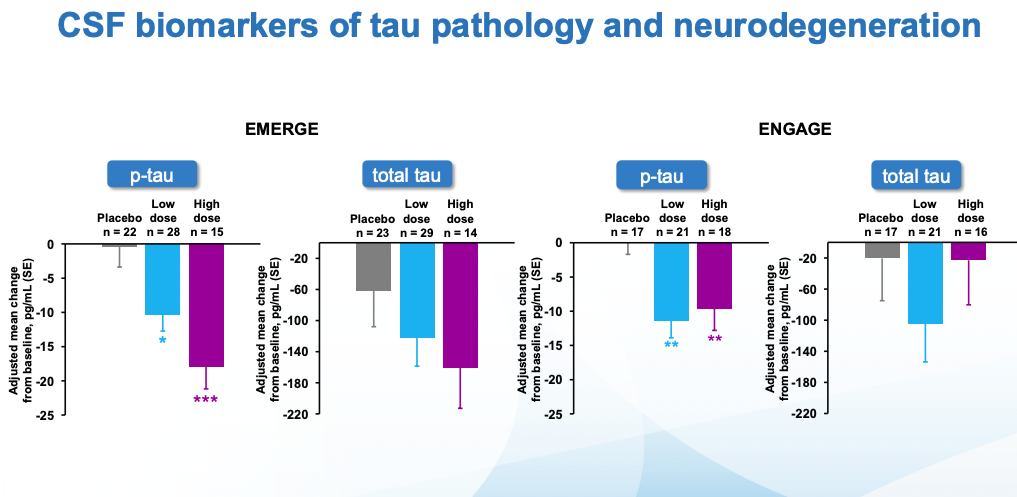
Cognitive scores kept getting worse in all cases. In ENGAGE, there was no difference wrt placebo. EMERGE is the interesting one, there one can see that the low dose, and specially the high dose are statistically different from placebo. What's more, looking at tau (which remember is what seems to be most causally linked to neurodegeneration), the seeming benefits of EMERGE go hand in hand with an increased reduction in tau (though note that the CSF biomarkers group is much smaller than the PET one)
These results were not free of side effects: In a dose-increasing manner, patients were substantially more likely to suffer from cerebral edema (ARIA-E) and hemorrhages (ARIA-H).
Then we get to some massaging of the data (depending on who you ask this is bad practice or something reasonable to do. I tend to like pre-registered studies), excluding outliers and narrowing the patient population to one that had received the highest doses. Applying these criteria to both studies one gets to the following (Note that the 78 weeks timepoint has substantially fewer patients, because most discontinued treatment when Biogen deemed the trials futile in March 2019 and discontinued them):
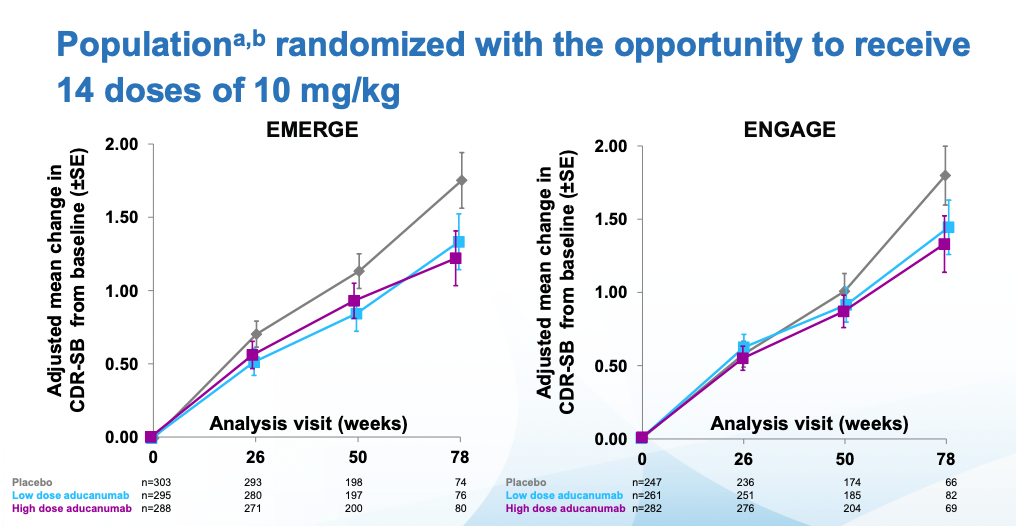
The FDA approved the drug after second view of the data. Crucially, they did not approve the drug based on effectiveness, they approved it based on the data that showed reduction in amyloid plaque (which it did reduce)
Immediately after the decision, many scientists wrote replies questioning the decision. Before going into that it's worth examining what the results, at face value, mean. By week 78, patients on the highest dose had gotten worse 30% less than placebo. Alzheimer's is a disease that progresses slowly. In those 78 weeks the placebo group moved two points on the CDR-SB scale which, again if you check my prior post you can see it goes up all the way to 18. Patients with Alzheimer's have on average a score of 7.4. Given the starting scores for placebo and treatment groups, they are not there yet. Longer followups would be warranted.
The most comprehensive examination of aducanumab is the Institute for Clinical and Economic Review (ICER)'s report . The report ultimately aims to answer whether aducanumab is cost-effective at the price Biogen intends to sell it. Whether aducanumab works and whether it's worth the price may seem like different questions: However, empirical measurements always have some noise, so we have to establish cutoffs for how certain we are that something "worked". These cutoffs (Think, for example p-values) should be established based on cost-benefit analysis. For example, if aducanumab completely stopped the progression of Alzheimer's but it costs one trillion dollars, then it would not be advisable for a public healthcare system to fund its use. Rather, it would be extremely valuable to figure out how to reduce its price, or to develop alternative drugs that achieve the same effects, which should be easier to do once the mechanism is known to work.
Here I am more interested in the "does it work" part of the equation, but in any case on ICER's model, aducanumab is not worth it unless one is particularly optimistic about the data and the drug gets a 80-90% reduction in price. You can read a summary of their reasoning here.
ICER's first critique is that the 22% slower reduction in cognitive function with aducanumab is so small that it's unclear if patients would feel any different. It's 22% better by a particular scale (CDR-SB), but patients may not agree that they improved function by 22% in the sense that they care about. An study they cite (Andrews et al., 2019) tries to establish thresholds for clinical significance for CDR-SB: these depend on how advanced the disease is. It's easier to tell if someone's cognitive function transitions from unimpaired to mild AD than to see if someone already deep into AD is getting even worse. In the case of mild AD (aducanumab's target population), the threshold they ballpark is a change in 1.63 points. The change observed in the high dose arm of the EMERGE trial (Which was statistically significant) was just 0.39 in favor of aducanumab.
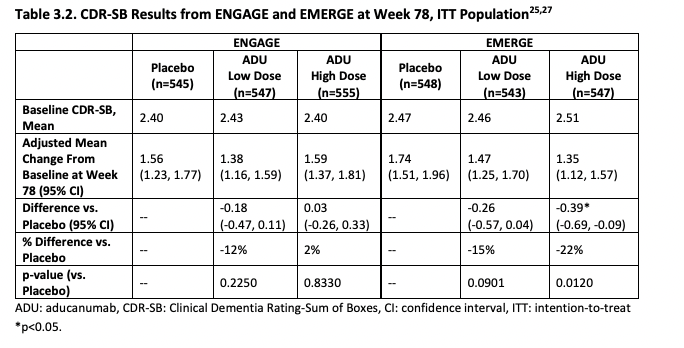
ICER then pools EMERGE and ENGAGE together (They study designs were similar enough to allow this), finding that combined the low dose looks to more clearly be having an affect than the high dose:
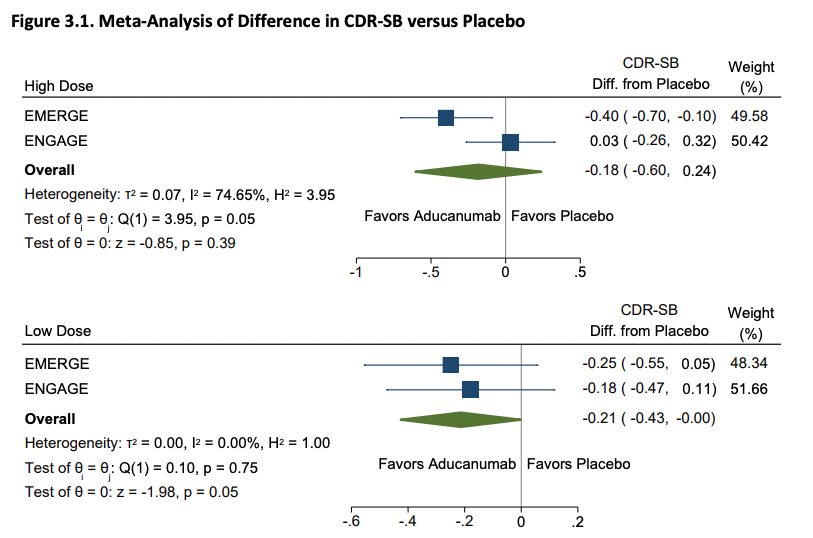
ICER then raises some doubts about Biogen's post-hoc analysis: Biogen had argued that the exposure to higher doses was unbalanced across the trials (which is true) and that this explains the result. This may be a bit confusing so here's a picture from the Biogen slides: The dosing scheme in the clinical trial depends on preexisting characteristics of a patient (chiefly apoE status) and the arm they are in.
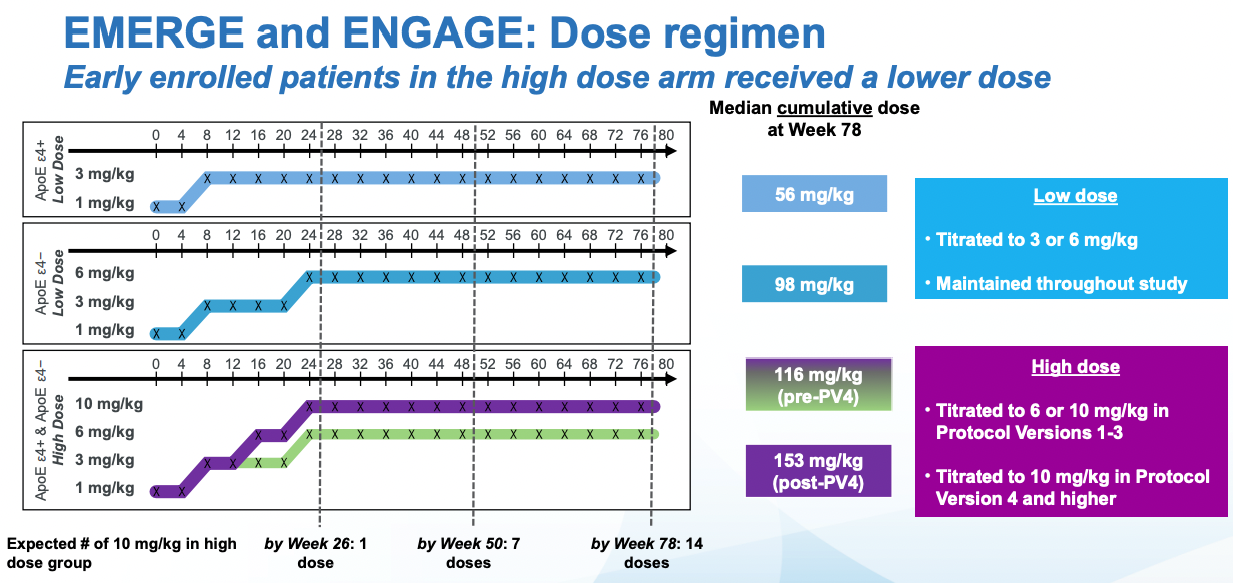
The dosing is not the same throughout the trial, it escalates. And if there are adverse events, one given participant may not get a further dose until the symptoms resolve. Given all of this, one way to analyze the data is with "Intention to Treat" (ITT) analysis. This is taking the results in the control and intervention group at face value: if participants leave they don't get excluded from the analysis (This may underestimate side-effects). If a patient receives a smaller dose in a particular timepoints because a higher dose causes side effects that may lead to a reduce final effect, but it keeps the effect estimate from the study closer to what one would observe in the real world (Where a doctor would stop treatment). Aducanumab is administered intravenously, but in trials where patients have to take pills, the patients may not take the pills (maybe they forget) and so part of the treatment group won't be exposed to the drug. These patients will still be in the treatment group (Reducing the effect of the intervention) if one is doing ITT analysis.
Splitting patients by the cumulative exposure to aducanumab does show that the results are similar in both trials at the highest dose. At an intermediate dose, the results if anything are discordant which is not what one would expect and might have been due to improper propensity score matching. The effects reported below are not just taking the observed effects and comparing them, they are controlling for all sorts of things like (from the Biogen presentation): effects of treatment group, categorical visit, treatment-by-visit interaction, baseline CDR-SB, baseline CDR-SB by visit interaction, baseline MMSE, Alzheimer’s disease symptomatic medication use at baseline, region, and laboratory ApoE ε4 status. It would take longer to properly assess the effect of all of these on the outcome, but suffice to say that in my mental model of data credibility, both post-hoc analysis and extensive statistical controls in a RCT context both decrease my confidence that the result is right, and both the FDA statistical reviewers and ICER thinks so as well.
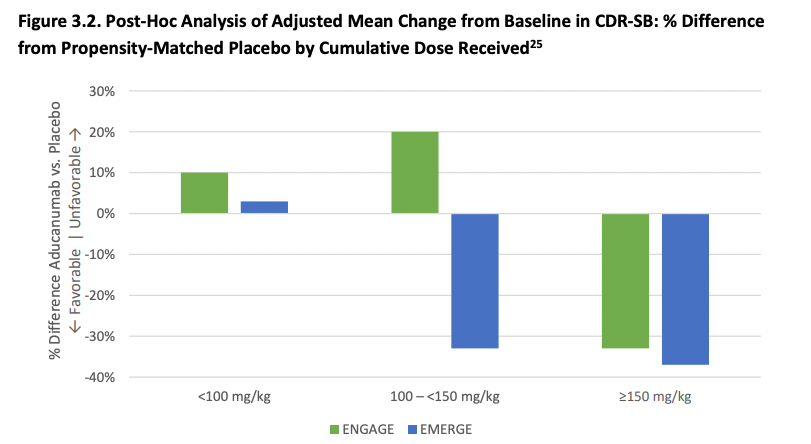
Next, ICER argues (based on FDA reasoning), if aducanumab works but only at a higher dose then we would expect that various subgroup analyses to also show a benefit that increases with dose, but this is not obvious at all from the data and is compatible with the placebo group having gotten worse faster (due to chance) in the ENGAGE trial. And then of course, the low-dose group of ENGAGE had better outcomes than the high-dose group also of ENGAGE which is compatible with noise and not as much with the high dose being the key to a successful treatment. As was the case with the FDA advisors, ICER's 15-person appraisal committee disagreed that there is adequate evidence that on net aducanumab is better than supportive care.
No one that has looked at the data disputes that higher doses of aducanumab remove more amyloid plaque and also lead to more side effects. The side effects in the short term are easier to measure than the long-term effects on cognition of aducanumab and they factor in in ICER's all things considered analysis. These side effects do not seem to be key drivers of their final cost-benefit analysis.
ICER gave external parties, including Biogen or the Alzheimer's Association, the opportunity to review drafts of their report and provide feedback. Nothing in what they say represent valid criticism to me. In fact, at worst they represent examples of statistical illiteracy on the side of the patient associations. I'll leave you with one such critique, and ICER's reply:
AA: In its effort to evaluate the cost effectiveness of aducanumab, ICER assumed blended efficacy of the ENGAGE and EMERGE trials. We dispute and question ICER’s approach. EMERGE met its prespecified primary outcome and found in the high dose aducanumab group a 22% reduction in decline on the CDR-SB--an outcome that was evident even under the situation of early trial cessation. The argument made by ICER that “the primary outcome of CDR-SB, while a validated scale, is not used frequently in clinical practice and thus the minimal clinically important difference has not been established” is misconstrued.
ICER: While we appreciate that there are differing views on how to interpret the discrepant results between ENGAGE and EMERGE, there is no a priori reason to believe the results of one trial over the other. The scientific method begins by assuming an intervention has no effect (no harms and no benefits), also known as the null hypothesis. Conventional scientific approaches place the onus on an intervention, through evidence generation and corresponding analyses, to demonstrate alternatives to the null. Biostatistics, epidemiology, and pharmacoeconomic good practices all argue for best-available evidence approaches in assessing an intervention’s benefits and harms. As the Evidence Report communicates, we support approaches that synthesize evidence across all comparable trials to quantify benefits and harms of aducanumab. In the case of aducanumab, it is as likely that the results from ENGAGE are true (and perhaps more likely given prior failures of drugs in this class) as it is that the EMERGE results are true. Blending the results seems like the fairest approach in this situation, though we recognize that the true effect of aducanumab may not be the average of the results of the two trials. Furthermore, we present scenarios that focus on the results of EMERGE being true; if, in fact, the results of ENGAGE are true, then the therapy has no value. With respect to the CDR-SB, our review of the literature and discussion with multiple experts revealed no consensus on what a clinically relevant difference in the scores would be, and several experts had concerns that the differences seen in EMERGE were too small to be clinically meaningful.
Solanezumab: This happened before
Pharma has ran clinical trials for a number of other monoclonal antibodies before aducanumab. Aducanumab is also not the first case where the trial sponsors try to aggressively massage their data to get their drug approved. Solanezumab could be a model for what could happen with aducanumab in future trials (Knopman et al., 2020):
Recent experience with solanezumab should be a cautionary reminder of the fickleness of claims based on post hoc analyses. After a pair of phase III trials failed to achieve their goals, review of the unblinded data suggested that mild, but not more advanced, patients benefited from solanezumab.8 These observations spawned a third trial, EXPEDITION 3, that was restricted to mild AD dementia. Unfortunately, it too failed to meet its goals.7
Solanezumab is now universally acknowledged to have failed, so it is instructive to, with the benefit of hindsight, look at how data can be made to look promising post-hoc while at the same time a trial designed to confirm those results can fail in a clear way.
The paper presenting the results from the two original trials is Doody et al. (2014) and the breakdown into mild Alzheimer's vs moderate looks as follow:

So you could look at this and say well the primary endpoint were the ADAS and ADCS scores, those have lower p-values in the mild AD group so... maybe there's something to that particular patient population? To me this looks very weak sauce, but the lure of billions of dollars in revenue was enough to merit one more trial that was a clear failure:
Honig et al. (2018)
in prespecified pooled secondary analyses, patients with mild Alzheimer’s disease who were treated with solanezumab had less cognitive decline by approximately 34% and less functional decline by approximately 18% than did patients who received placebo.6 We report the results of a third double-blind, placebo-controlled phase 3 trial (EXPEDITION3), which enrolled only patients who had mild Alzheimer’s disease, defined as a Mini–Mental State Examination (MMSE) score of 20 to 26 (on a scale from 0 to 30, with higher scores indicating better cognition), and had biomarker evidence of cerebral beta-amyloid deposition. [...]
In conclusion, in patients with mild Alzheimer’s disease, the results of the EXPEDITION3 trial showed no benefit of solanezumab on the primary outcome of cognitive decline and did not reproduce the secondary analyses of the EXPEDITION and EXPEDITION2 trials. The rationale for further trials with solanezumab with different doses and timing may require examination.
Lecanemab: Is there any hope?
As I mentioned in the aducanumab section, lecanemab, the latest in the series of monoclonal antibodies against amyloid, targets a different kind of amyloid aggregates. Lecanemab recently showed some promise in a press release from Biogen where they announced results from their phase III CLARITY AD study:
- Compared to placebo, a slower cognitive decline (27%, or 0.45 points in the CDR-SB scale)
- Compared to the aducanumab trial, substantially reduced incidence of side effects, particularly brain edemas and microhemorrhages (ARIA-E and ARIA-H)
- Injections of lecanemab twice a week
- A patient population with mild cognitive impairment and mild AD with confirmed amyloid pathology
- A followup for 18 months
Unlike aducanumab or solanezumab, these are primary analysis results, following straight from the pre-registered analysis plan, hence these results are more credible. However, even when taken at face value, the clinical relevance of that 27%, which is similar to the optimistic interpretation of the aducanumab results, is unclear. ICER will be publishing an evaluation of lecanemab after Biogen published more data in November, but I suspect their assessment won't be particularly optimistic. It does look better than aducanumab, and given the reduced odds of side effects, there may be a lower price at which it is cost effective. Lecanemab has, for now, some chance of uncontroversially showing that amyloid removal does something good for patients, even if it is far from stopping Alzheimer's, but all things considered it won't be worth the cost and hassle (biweekly injections and side-effects).
All the trials, everywhere, all at once
I have only discussed three of these monoclonal antibody trials for amyloid, but I am not cherrypicking here: They really are illustrative of the general trend.
Willy Chertman send me this comment from someone that has been working on Alzheimer's dor a decade. It is illustrative
As someone who was in the trenches of Alzheimer's drug development since 2010, this is just unfathomable.
I watched as semagacestat, bapineuzumab, solanezumab, gantenerumab, crenezumab, verubecestat, lanabecestat, atabecestat, umibecestat [these last -becestats are b-secretase inhibitors], and elenbecestat all fall in that sequential order.
I was at AAIC when Biogen presented the aducanumab PRIME Phase 1b data and let me tell you, the room did not believe it. I have been to countless neurology conferences and watched blood trying to be rung from a stone.
No one had spent more money on anti-amyloid beta antibodies than Eli Lilly, and EXPEDITION 3 was an unmitigated disaster. Even after they pooled EXPEDITION 1 and 2 and analyzed them, they then just replicated that patient population in EXPEDITION 3 and failed to find any shred of efficacy.
Once of the Alzheimer's drugs I was working on was declared futile in Phase 3 the day I closed on my new house, and I have the picture of my, my wife, and our newborn in my hands just hours after I got the call.
I don’t know if anymore wants these drugs to work more than me because I have both personal and professional connections to some of them, but I refuse to bend clinical science and medicine “because shit is hard the last 20 years”.
You can read more here.
Primary prevention of Alzheimer's
The standard answer from the AD world to the repeated failures of these drugs is we need to intervene earlier. This is also an approach to therapu suggested by the modified ACH (where tau is a prion-like disease that becomes self-sustaining). Primary prevention is a term that refers to preventing the disease from ever happening in the first place. This requires intervening very early.
The only trial that is planning to do this is the DIAN-TU trial. It may be very expensive to run this trial in the regular AD population, so the trial is restricted to patients with mutations that deterministically cause AD. This trial hasn't finished yet, so in the meantime we could look at data of trials that have intervened early, but not as early.
One such trial is the DIAN-TU-001 (Salloway et al., 2021), in a patient population that is young (for an AD study, 42-26 years on average), presenting with dominantly inherited AD (DIAD, having mutations that deterministically cause AD). Participants were assigned to a placebo arm, gantenerumab, or solanezumab. This was not a very large trial, only about 30-40 patients completed each arm. Whereas the aducanumab trials followed patients for less than two years, DIAN followed them for four. The trial failed: as was the case for every other trial, cognitive function declined similary in intervention and control groups. Amyloid plaques were removed as one would expect. Unlike some of the other trials, this one also inclued FDG-PET (that measures glucose consumption in neurons, the most direct proxy of activity or at least presence or absence of neurons). Consistent with neurodegeneration, cognitive function, and glucose consumption being tightly coupled together, FDG-PET showed no difference between treatment and control.
Analysis of clinical trial data should not engage in naive frequentism
If you run 100 RCTs of the same drug against the same target, it is highly likely that 5 will show some effect even if there is none. Under the current FDA framework there is nothing wrong with this and those 5 ineffective drugs will get approved. On paper, one can point to the EMERGE trial and get the drug approved, and act as if ENGAGE never happened.
If a single paper ran 100 such experiments, reviewers would be asking them to correct for multiple comparisons. This seems to be forgotten when one assesses entire literatures rather than single pieces of work. From time to time one can find researchers making this exact same point, but when it comes the time to analyze the next paper, this is rarely taken into consideration. A rare example is this piece of Peter Bach at Stat News where he makes the exact same point I'm making here. Vinay Prasad also makes the same point in the context of oncology.
Better than say doing Bonferroni corrections, one should rather try to do a more Bayesian analysis where the data for a given trial is examined on the light of prior data, from other similar, even if not fully identical trials, across companies and labs. This is what we are all doing when intuitively (but rigorously and reasonably) analyzing data anyway. On paper, aducanumab, solenuzemab, or lecanemab are different, so we can't just naively pool the data, but we can make some assumptions based on their putative mechanisms (We can guess what to expect in terms of effect size from previous experiments that remove amyloid plaques even if a particular antibody targets a different kind of amyloid). If you asked me whether the next monoclonal antibody against amyloid beta will clearly succeed in having an uncontroversial clinically meaningful effect I will tell you no it won't (and I will be you on that). If you ask me about targeting inflammation or anything else that's way less explored I am less confident: maybe there is something to those.
Does this falsify the Amyloid Cascade Hypothesis?
These trials are still compatible with some form of the Amyloid Cascade Hypothesis. What I think is going on is that amyloid beta on its own has some negative effects that are being counteracted by the mAbs. This effect is probably real, but very minor. Under the revised form of the ACH I discussed in my introductory post, targeting amyloid once AD started is useless: the disease at that point is self-sustaining, and the results observed in the clinic are compatible with this.
What would not be compatible with this modified ACH?
- Monoclonal antibodies administered prophylactically decades before AD starts should work. At the very least, clinical trials for mAbs targeting amyloid beta should focus exclusively in patients that have at worst mild AD but ideally are just healthy, maybe in their 50s. If this doesn't work it would be significant evidence that the ACH is false in humans.
- Pervasive removal of tau and amyloid, at the same time should lead to a substantial slowdown or even stop the progression of the disease.
These two tests have not yet been done in vivo. Doing (1) is expensive, and there is seemingly only one such trial ongoing, the DIAN-TU Primary Prevention trial. It will be a few years before we have this data. But what about 2?
Number two for some reason doesn't get as much publicity as the amyloid-targeting mAbs but arguably they should get more, given that in the ACH tau is causally closer to neurodegeneration. There are a number of tau mAbs in the pipeline (Cummings et al., 2022). One of those have already failed in phase 2 studies: Semorinemab (Teng et al., 2022). But this is just the first trial ever of a tau-targeting therapy. Given it's a hitherto untouched target, I regard it as more promising than anti-Ab mAbs. Of course perhaps to effectively break Alzheimer's feedback loop one needs to target amyloid beta and tau at the same time. There are currently no ongoing trials doing this in humans.
Hence for now the ACH is still a feasible model for what's going on in the AD brain. What has been shown to be bunk is a model where Ab is the constant driver of neurodegeneration. The current data is compatible with Ab being a spark and tau being the engine, as well as a secret, more complex third thing (perhaps inflammation, infections, glymphatic system impairments, etc) being another key driver. I will review these in the next post.
Changelog
- 2022-10-18: Tim Peterson pointed me to the DIAN trial which got me to write the Primary Prevention section
Citation
In academic work, please cite this essay as:
Ricón, José Luis, “The failure of monoclonal antibody therapy for Alzheimer's Disease, implications for the Amyloid Cascade Hypothesis”, Nintil (2022-10-17), available at https://nintil.com/alzheimers-clinical-trials/.
Comments